BCM-514
Biochimie des protéines
6.0 Analyse des protéines purifiées
6.2 Spectrométrie de masse
Voici une adresse fort utile pour être initié aux principes de la spectrométrie de masse par le Dr Alison Ashcroft de l'université de
Leeds
Vous ne pouvez pas vous tromper non plus en visitant le site de l'université de l'Arizona.
En un mot, un spectromètre de masse c'est une balance. Il ne fonctionne pas du tout comme une balance, mais c'est tout de même ce qu'il est. Vous lui donnez un échantillon, et il va vous donner un poids -mais d'une manière vraiment précise... Pour une protéine de 40 000 Da, le spectromètre de masse peut vous indiquer un poids qui n'aura une marge d'erreur que de 4 Da!!! C'est amplement suffisant pour détecter le changement d'un acide aminé en un autre, ou encore la présence d'un groupement hydroxyl supplémentaire.
Mieux encore, si vous avez accès à une information génomique complète (comme c'est le cas pour de plus en plus d'organismes dont les génomes ont été séquencés) vous pouvez utiliser le spectromètre de masse pour identifier des protéines. Ainsi, si vous digérez la protéine XYZ (encore inconnue) à la trypsine et que vous en analysez les fragments par spectrométrie de masse, vous recevrez une liste de poids moléculaires. Un ordinateur peut facilement comparer les poids de cette liste avec les poids de toutes les séquences polypeptidiques codées par les cadres de lecture du génome qui nous intéresse, et identifier celles qui coïncident. Quand vous entendez un biochimiste dire qu'il a découpé une bande d'un gel SDS et l'a envoyé en mass spec, c'est tout juste ce qu'il a fait.
Il existe plusieurs types de spectromètres de masse. Ils portent des noms comprenant des sigles ésotériques comme TOF (time of flight), MALDI (matrix assisted laser desorption), ESI (electrospray ionisation), APCI (atmospheric pressure chemical ionisation), EI (electron impact), CI (chemical ionisation), FAB (fast atom bombardment), FD/FI (field desoprtion/ field ionisation), TSP (thermospray ionisation), Q (quadrupole), FT-ICR (Fourier transform-ion cyclotron resonance) etc.; ces sigles sont souvent combinés entre eux pour refléter la nature de l'appareil ou ce à quoi ils sont connectés (LC-MS = liquid chromatography -mass spectrometer, etc).
Le principe général de la spectrométrie de masse repose sur une ionisation du matériel et au voyage du matériel chargé vers un détecteur. On distingue plusieurs systèmes de spectrométrie de masse selon la technique d'ionisation et la technique de détection.
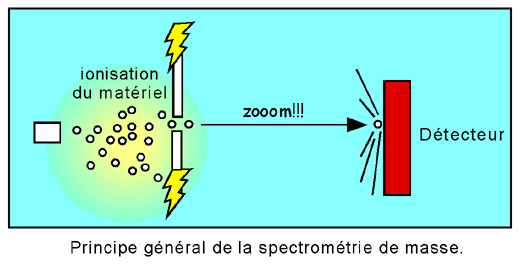
6.2.1 Ionisation
6.2.1.1 EI (electron ionization, jadis electron impact)
Ici, on utilise la chaleur pour faire perdre des électrons à un filament de tungstène ou de rhénium dans lequel passe un courant électrique. Ces électrons sont emportés par un champ magnétique induit par un aimant, ce qui cause la formation d'un faisceau d'électrons avec lequel les particules gazéifiées de notre échantillon vont entrer en contact. L'impact va briser les protéines en plus petits peptides et les ioniser.
À basse énergie (environ 20 eV), les électrons ne résussissent pas à provoquer une ionisation. À environ 70 eV, le transfert d'énergie entre les électrons et les molécules organiques analysées est maximisé, ce qui favorise l'ionisation des peptides.
6.2.1.2 FAB (Fast atom bombardment)
Ici, c'est un faisceau d'atomes de gaz (argon ou xénon) de haute énergie (4-10 KeV) qui bombarde la substance à analyser. Cette énergie est transmise à nos peptides qui sont ejectés sous forme ionique (positive et négative). Si la source d'accélération (les deux éclairs dans la figure ci-haut) induit un courant positif, ce sont les ions négatifs qui seront accélérés vers le détecteur; si le courant induit est négatif, ce seront les ions positifs.
6.2.1.3 ESI (electrospray)
Ici, l'échantillon est d'abord soumis à un fort champ magnétique pour en ioniser les molécules. Celles-ci sont pulvérisées dans la chambre d'ionisation sous la forme de microgouttes pleines d'ions (comme si elles sortaient d'un pouiche-pouiche pour arroser les plantes), gouttes qui sont rapidement déshydratées par un courant de gaz chauffé (il s'agit le plus souvent d'azote). Le solvant disparaissant, les ions de même charge dans les gouttes finissent par ne plus pouvoir se supporter, la faisant exploser en plusieurs gouttelettes plus petites. Le processus se répète jusqu'à ce qu'il n'y ait plus de solvant du tout, laissant des ions "nus" dans les airs.
Les charges sont distribuées sur les peptides de façon statistique; un même peptide peut en porter plusieurs. Un séchage plus rapide des gouttelettes favorise la multi-ionisation.
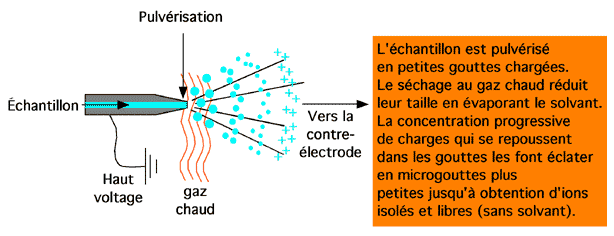
Une sectrométrie de masse utilisant l'ESI présente souvent ses résultats sous forme de m/z (ou masse / charge) pour présenter la proportion des différentes formes chargées d'un même peptide.
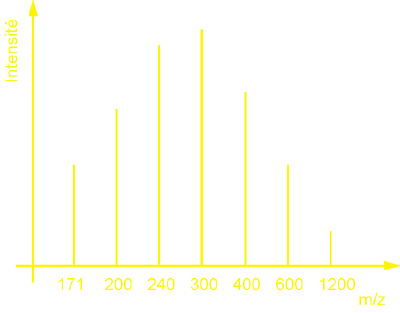
Le graphique ci-haut présente la distribution des ions détectés par un spectromètre de masse et distribués selon leur ration m/z. Comme la valeur de z (la charge) ne peut être qu'un nombre entier, il devient assez facile de déterminer quelle était la masse du peptide initial qui a fourni tous les ions. Dans ce cas-ci, on devine que ce peptide avait une masse de 1200 (car 1200/2=600; 1200/3=400; 1200/4=300; 1200/5=240; 1200/6=200 et 1200/7=171 et des poussières). Si l'ionisation avait été particulièrement efficace et qu'on n'avait pas eu d'ions monovalents, le pic m/z de 1200 n'aurait pas été là. Malgré cela, on aurait quand même pu déterminer que le peptide initial avait une masse de 1200 en identifiant le ppcm (le plus petit commun multiple, comme on disait à la petite école!) de 171, 200, 240, 300, 400 et 600.
Le site suivant explique l'interprétation d'un spectre d'ESI.
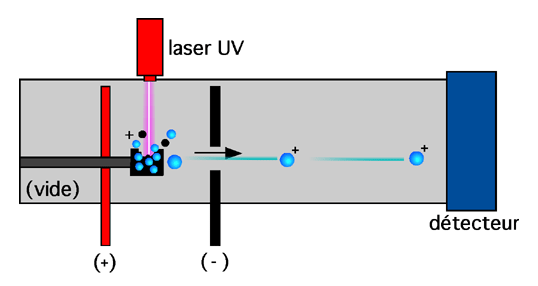
6.2.1.4 MALDI (matrix-assisted laser desorption ionisation)
Ici, l'échantillon est d'abord associé à une matrice de telle façon que chaque molécule de peptide soit séparée de ses voisines. Les peptides et la matrice doivent former une "solution solide" (un mélange qu'on a laissé sécher jusqu'à ce qu'il forme un biscuit, en quelque sorte!) dont tout le liquide a été retiré. L e s composés les plus populaires pour la composition de la matrice sont l'acide sinapinique, l'acide alpha-cyano-4-hydroxycinnamique et l'acide 2,5-dihydroxybenzoïque. Ces molécules ont un poids moléculaire assez faible pour permettre leur vaporisation quand on les excite avec assez d'énergie, mais aussi assez élevé pour prévenir leur évaporation spontanée pendant ou après la préparation de l'échantillon. Elles sont en outre acides, ce qui facilite la protonation des peptides, et absorbent la lumière UV (ce qui joue un rôle crucial dans l'étape suivante).
L'échantillon est ensuite zappé avec un laser UV. L'énergie du laser est absorbée en quelques nanosecondes par la matrice qui chauffe très rapidement. Comme les peptides de l'échantillon sont associés à un nombre bien plus grand de molécules de la matrice, ce sont ces dernières qui absorbent le gros de l'énergie; les molécules peptidiques ne sont donc pas endommagés par le laser. Cette excitation énergétique a plusieurs effets. Elle cause d'abord la désoprtion de certains peptides qui sont expulsés. De plus, le transfert subit d'énergie de la matrice aux peptides peut les ioniser. (On peut observer des transferts de protons de la matrice vers les peptides; on peut aussi voir les peptides perdre des électrons ou en gagner. Le choix de la matrice joue un rôle important sur l'état ionique final de l'échantillon).
Comme toute cette opération se déroule entre une anode et une cathode, les ions sont ensuite propulsés en direction de l'électrode portant une charge inverse à la leur -et vers un détecteur. (Les peptides peuvent, mentionnons-le, porter de multiples charges; quoique dans le cas du MALDI, les peptides n'en portent d'habitude qu'une seule).
6.2.2 Détection
6.2.2.1 TOF (time of flight)
Cette technique prend en considération le temps que met un ion pour parcourir le chemin de la chambre d'ionisation au détecteur. Les ions plus gros mettent plus de temps que les plus petits.
Elle ne semble pas limitée par la taille des ions et est extrêmement précise. Elle est souvent combinée avec une méthode d'ionisation rapide comme le MALDI, qui permet de savoir exactement quand les particules ont pris leur envol à cause de la vitesse du processus MALDI (un rayon laser voyage en effet à la vitesse de...ah, vous le saviez déjà).
6.2.2.2 Analyse par double focus
Cette technique (aussi appelée sector analysis) permet de focuser les ions de même charge et de même masse mais ayant un niveau d'énergie différent, ce qui augmente la résolution de la détection des ions de même nature.
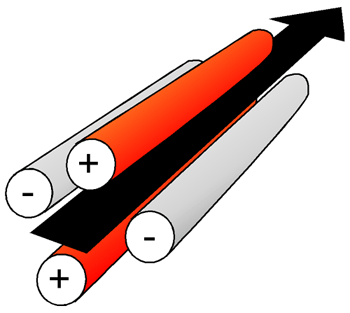
6.2.2.3 Q (quadrupole)
|
un spectromètre Q utilise des champs électriques pour séparer les ions selon leur masse et leur charge. Le quadrupole consiste en quatre fils parallèles formant les arètes d'un quadrilatère: deux sont positifs, deux négatifs.
|
|
Dépendant des champs électriques produits avec le courant passant dans les fils, seuls les ions avec un ratio m/z particulier se rendront jusqu'au détecteur au bout du quadripole. Les autres seront déviés sur les fils. En variant la force et la fréquence des champs, différents ions seront détectés, nous donnant ainsi leur spectre de masse. Ne vous fiez pas à la grosse flèche droite de la figure, qui ne vous indique que la direction générale. La course d'un ion dans un quadripole ressemble plus à celle d'une abeille saoûle.
6.2.2.4 FT-ICR (Fourier transfrom ion cyclotron resonance).
Ici, un piège à ions dans un champ magnétique statique et uniforme dans l'espace force les ions à adopter une orbite circulaire dans ledit champ; la fréquence de cette orbite dépend de la masse, de la vitesse et de la charge de l'ion. Des détecteurs suivent la course des ions et permettent d'en déterminer ces paramètres.
6.2.2.5 Piège à ions (ion trap)
Dans ce piège à ions, un autre type de quadrupole est capable de capturer les particules ionisées selon leur masse et leur charge, ainsi que selon le courant qu'on fait passer dans le quadrupole. C'est la trajectoire des ions capturés dans des conditions données de potentiel au niveau du quadrupole qui donne les informations sur m et z.
6.2.3 Autres techniques de spectrométrie de masse
6.2.3.1 MS/MS ou MSn
Cette technique commence par séparer les ions en les faisant passer à travers une série de trois quadrupoles qui sont réglés pour ne laisser passer que certains ions vers le détecteur (par exemple, un certain peptide tryptique). Un MS laisserait normalement passer tous les peptides issus d'une digestion protéique, et nous donnerait le poids de chacun. Dans le MS/MS, on commence par n'en laisser passer qu'un.
Le peptide qui réussit à passer entre dans une chambre où il est frappé par des molécules de gaz chargées qui brisent sa chaîne peptidique en un ou plusieurs endroits. Ces bris font en sorte que ce sont des morceaux du peptide tryptique qui finissent par arriver au détecteur. En sachant le poids théorique du peptide tryptique au départ et en comparant les poids des différents petits peptides qui en sont issus, on peut établir l'ordre des résidus contenu dans le fragment initial. Cette technique de MS/MS permet donc de séquencer un petit peptide.

6.3 Dichroïsme circulaire
La spectroscopie par dichroïsme circulaire nous permet d'étudier la structure secondaire (hélices, feuillets, etc.) de polypeptides et de protéines en solution. Cette technique utilise peu de matériel et ne le détruit pas. Les changements dus aux conditions ambiantes de pH, d'agents dénaturants et de température peuvent y être facilement monitorés.
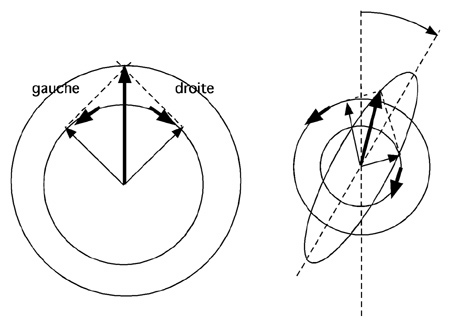
La technique repose sur la capacité qu'on les structures optiquement actives d'absorber de façon inégale la lumière polarisée circulairement à droite de la lumière polarisée circulairement à gauche.
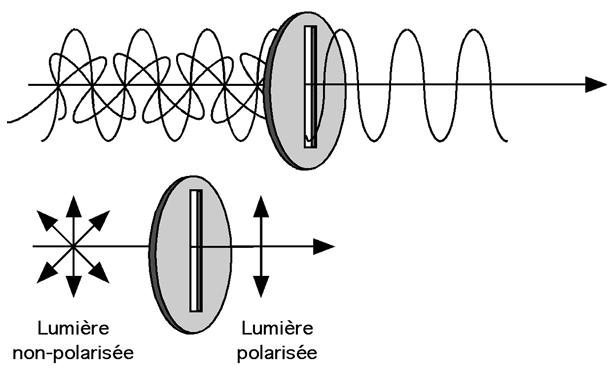
Une minute... "lumière circulairement polarisée?" vous demandez-vous. Qu'est-ce que c'est encore que cette diablerie?
| Vous êtes probablement familiers avec la théorie ondulatoire de la lumière. Une onde lumineuse d'une certaine amplitude voyage dans une direction donnée avec une forme sinusoïdale. L'angle des pics de l'onde par rapport à l'axe de sa propagation peut avoir n'importe quelle valeur, mais il est possible de sélectionner un angle particulier grâce à un filtre polarisant (polaroïd). Consistant en de longues molécules alignées dans une direction précise, un tel filtre ne laisse passer que les oscillations d'une onde électromagnétique orientées de même façon (dans son axe de transmission). |
|
Maintenant, si nous combinons deux rayons lumineux polarisés à 90° l'un de l'autre mais se propageant dans la même direction. On obtiendra les ondes suivantes:
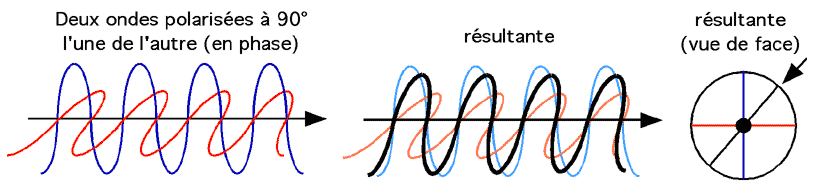
La résultante des deux ondes est une autre onde sinusoïdale située entre les deux premières. Chaque pic de l'une correspond au pic de l'autre, et chaque noeud de l'une arrive au même point que pour l'autre. Mais si notre filtre (un modulateur photoélastique), en plus de laisser passer ces deux ondes, les déphase en plus d'une valeur de π/ 2, le résultat sera tout autre. La résultante sera une spirale.
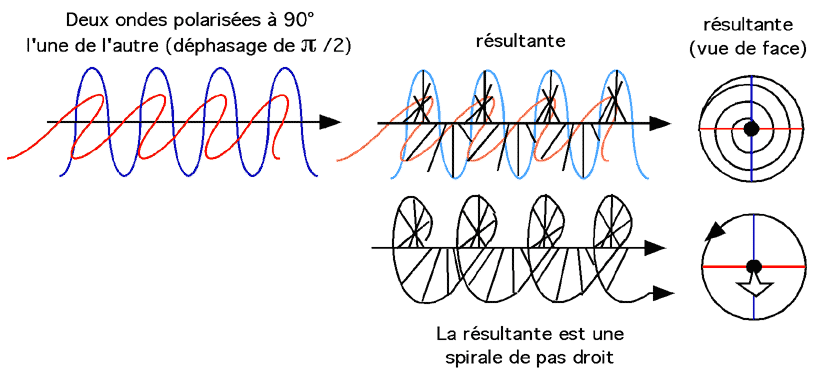
Dans L'exemple ci-dessus, on a retardé l'onde verticale bleue d'un quart de longueur d'onde (λ / 4, ou encore π / 2 radiants) pour une résultante en hélice de pas droit. Si on avait plutôt avancé l'onde bleue de π / 2 par rapport à la rouge, on aurait obtenu une hélice de pas gauche. Pour une démonstration animée, veuillez vous référer à l'excellent site
suivant.
Les structures secondaires n'absorbent pas de façon égale la lumière polarisée circulairement vers la droite et la lumière polarisée circulairement vers la gauche. On raisonnera que l'absorption préférentielle de l'une de ces deux polarisations résultera en une déviation de la résultante (au lieu d'un cercle, le tracé de la résultante entre la spirale tournant à droite et la spirale tournant à gauche donnera une ellipse).
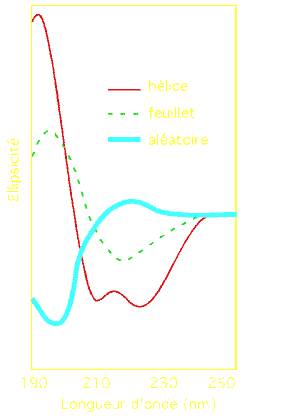
Cette déviation est appelée dichroïsme circulaire. Elle permet d'établir un spectre (appelé CD spectra en anglais) de forme caractéristique pour trois structures secondaires retrouvées dans les protéines: l'hélice, le feuillet et la forme aléatoire (ou random coil), qui chacune présentent des asymétries structurales.
Le spectre CD est particulièrement approprié pour juger rapidement de l'état de repliement d'une protéine (ses hélices sont-elles bien hélicales comme prévu?); pour comparer la structure de protéines obtenues de sources différentes; pour analyser l'impact de mutations ponctuelles surt la structure d'une protéine; pour juger de la stabilité de la structure face à des changements environnementaux (pH, salinité); pour déterminer l'impact sur la structure de la présence d'un cofacteur comme un le zinc ou la magnésium.
On peut aussi suivre au fur et à mesure qu'il se déroule le déploiement d'une protéine au fur et à mesure qu'on la soumet à un traitement dénaturant.
|
La forme du spectre obtenu pour une protéine peut être décomposée en ses trois composantes:
(a) hélicale,
(b) en feuillet,
(c) aléatoire.
Chaque composante donne une courbe spectrale caractéristique.
(Voir le site
suivant pour plus d'information.
|
La stabilité thermique d'une protéine peut être évaluée en suvant le comportement de sa structure à différentes températures; cette opération peut aussi se faire de façon continue avec un spectre de températures à une longueur d'onde fixe.
La dénaturation d'une protéine risque d'être irréversible. Le CD nous permet de la suivre "en direct", et de juger si elle est encore réversible. Trouver les conditions dans laquelle une protéine peut plus facilement retrouver son état premier après une dénaturation partielle peut nous être d'un grand secours pour la conserver longtemps.
6.4 Spectroscopie par Résonance Magnétique Nucléaire (RMN)
On utilise la RMN pour étudier la structure de petites protéines (jusqu'à 30 KDa environ) en solution sans avoir besoin à les cristalliser. La RMN utilise le spin des particules subatomique pour nous renseigner sur la structure des protéines.
Les électrons, les protons et les neutrons peuvent être imaginés comme des sphères tournant sur leur axe. Dans plusieurs atomes comme le carbone 12, de telles révolutions (le spin) sont appariées de telle façon que le spin global du noyau est égal à zéro. Dans d'autres atomes, cependant, comme le 1H ou le 13C, le noyau possède un spin.
(a) Si le nombre de protons et le nombre de neutrons sont tous les deux pairs, le noyau n'a pas de spin.
(b) Si le nombre de protons plus le nombre de neutrons est impair, le spin a une valeur qui finit en 1/2 (comme 1/2, 3/2, 5/2...)
(c) Si le nombre de protons et de neutrons sont tous les deux impairs, le spin a une valeur entière (1,2,3...)
Le spin global du noyau est noté I (i majuscule) et un noyau a 2I + 1 orientations possibles. Ainsi, un noyau de spin 1/2 possède (2 x 1/2) + 1 orientations, ou plus simplement 2 orientations. En l'absence de champ magnétique, ces deux orientations ont le même niveau d'énergie. Dans un champ magnétique, par contre, ces deux énergies vont diverger: le spin va soit être enligné dans le sens du champ magnétique (choix de basse énergie), ou lui être opposé (choix de haute énergie).
On peut faire transiter un noyau de l'état d'énergie faible à l'état d'énergie élevé en l'irradiant avec une fréquence donnée (dont la valeur exacte a à voir avec la précession du spin, mais là nous entrons dans des considérations de physique quantique qui, pour toutes intéressantes qu'elles puissent être, nous éloignent un peu de notre propos). Le retour subséquent du noyau à son état d'énergie antérieur est enregistrée par un détecteur. Ce phénomène de transition d'un niveau d'énergie à un autre est la résonance qui donne le R de RMN.
L'environnement des noyaux influencera la façon dont ils se comportent quand on les irradie à la longueur d'onde appropriée; la RMN permet donc de déterminer la nature des liens entourant un atome.
Pour en savoir plus long, n'hésitez pas à vous référer à ce site.
6.5 Diffraction aux rayons X
6.5.1 Principe
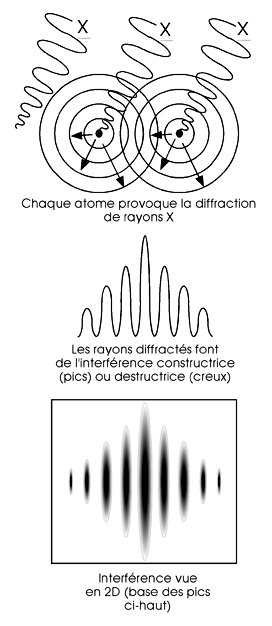
Dans un cristal parfait, toutes les sous-unités ont la même conformation et la même orientation. Quand des rayons X sont focusés à travers un cristal de protéines purifiées, ils sont déviés selon un patron particulier qu'on appelle le patron de diffraction.
Le nombre d'électrons dans les atomes de la protéine détermine le degré de diffraction des rayons, et c'est pourquoi les atomes lourds seront plus efficaces pour faire dévier les rayons X.
|
Chaque atome peut induire la diffraction de rayons X incidents. Les rayons déviés se répartissent dans l'espace. Si plus d'un atome est présent, les rayons déviés par l'un et l'autre se feront de l'interférence; cette interérence peut être constructrice quand deux pics, ou encore deux creux, se superposent. Elle peut aussi être destructrice quand ce sont un pic et un creux qui se superposent.
La succession d'interférence constructrices et destructrices donne le graphique du milieu; vue en 2D elle donne la figure du bas, ci-contre.
En combinant le patron de diffraction d'un cristal obtenu à partir de différents angles, on obtient assez d'information pour nous aider à déterminer l'agencement des atomes de la protéine dans l'espace.
La cristallisation demande une grande quantité de protéine pure.
|
6.5.2 Cristallisation
On cristallise une substance en la laissant devenir sursaturée dans un solvant; en permettant au solvant de s'évaporer, par exemple, ou en abaissant la température d'une solution saturée. Pour une protéine, on peut aussi faire varier le pH ou la force ionique de la solution.
Si un noyau de cristallisation est présent, il peut permettre la croissance d'un gros cristal, qui consiste en la répétion ordonnée dans l'espace d'une structure particulière.
Dans la figure ci-dessous, l'agencement régulier des sous-unité de la première structure lui donne une apparence cristalline. Dans le second cas, l'empilement des sous-unités est désordonné et non-répétitif; ce n'est pas un cristal. Dans le dessin de Moritz Escher, à droite, on voit un personnage dévaler un escalier jusqu'à ce que la répétition ordonnée de son image donne au bas de l'oeuvre une allure très cristalline. Le personnage au bas des marches a en quelque sorte servi de noyau de cristallisation.
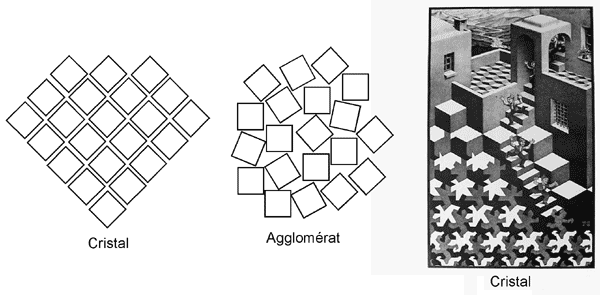
Un cristal de protéines, qui contient environ 50% de solvant réparti dans les interstices entre les molécules, tient ensemble par le biais de forces hydrophobes, de ponts hydrogènes et de ponts de sel (contrairement à un cristal minéral qui utilise surtout les liens ioniques). En fait de solidité, un cristal de protéine est donc plus proche d'un cube de jello sec que d'un diamant.
benoit.leblanc@usherbrooke.ca