BCM-514
Biochimie des protéines
5.0 La purification des protéines
5.3 Essai
Vous aurez compris à la lecture de ce qui précède que la purification est un processus étapiste. Une purification peut prendre plusieurs jours et nous voulons savoir à chaque étape où en est notre protéine.
Il est possible de vérifier la présence aussi bien que l'activité d'une protéine, le choix se faisant selon la nature de la protéine recherchée, la faisabilité du test et l'étape à laquelle nous en sommes dans la purification. Les premières étapes se contentent généralement de vérifier la présence de protéines (n'importe lesquelles) par spectroscopie; c'est plus tard pendant la manoeuvre qu'on commencera à se soucier de vérifier la présence d'une protéine particulière ou de son activité.
Quoiqu'il en soit, il est impératif de toujours, toujours garder des aliquots de matériel à chaque étape! De cette manière, on peut retracer après coup le déroulement de la purification, comprendre quelles en sont les étapes les plus utiles ou les plus dommageables, et améliorer notre protocole pour l'avenir.
5.3.1 Critères
|
Sensibilité
|
Parce que la protéine risque d'être très diluée à certaines étapes, particulièrement les premières.
|
|
Spécificité
|
Nous cherchons une aiguille dans une grosse boîte d'aiguilles, et comme rien ne ressemble plus à l'une d'entre elles que sa voisine, il vaut mieux que nous sachions vraiment reconnaître la nôtre.
|
|
Disponibilité des réactifs
|
Si votre protéine n'est qu'un des facteurs impliqués dans une réaction in vitro requérant huit autres protéines purifiées, mieux vaut déjà avoir ces derniers si vous comptez utiliser un essai réactionnel. Sinon, optez pour un essai que vous pouvez vraiment effectuer.
|
|
Coût |
Ce facteur se présente souvent.
|
|
Temps requis |
Plus votre purification est rapide et mieux ce sera. Votre essai devrait donc vous permettre d'obtenir rapidement une idée de ce qui se passe avec votre purification; si vous pouvez l'effectuer à chaque étape PENDANT la purification, encore mieux.
|
5.3.2 Exemples d'essais
5.3.2.1 Présence de protéines (en général)
(A) Par mesure de l'absorption aux UV.
Ce type d'analyse est très pratique pour suivre la course de protéines sur une série de colonnes de chromatographie. Tous les systèmes de chromatographie sont équipés d'une cellule spectrophotométrique pour mesurer l'absorption des rayons UV par le matériel qui sort des colonnes. C'est cependant une méthode simple dont le type de réponse est "oui, il y a des protéines" ou "non, il y a eu un os quelque part". On ne peut pas sérieusement s'attendre à pouvoir faire une relation exacte entre la densité optique d'un mélange protéique en cours de purification et sa concentration réelle. (On utilise parfois un coefficient d'extinction très approximatif pour dire qu'une densité optique à 280nm, dans une cuvette de 1 cm, correspond à 1 mg/mL de protéines...Mais c'est comme de dire que la population de la Terre s'élève à quelques milliards. C'est une idée générale, rien de plus).
En outre, n'oublions pas que l'ADN et l'ARN absorbent dans le coin de 250-260nm, ce qui risque de nous causer des soucis de précision! (D'habitude, on a plutôt le problème inverse, celui des protéines qui contaminent notre ADN. Mais tout finit par nous arriver si on vit assez longtemps).
On peut utiliser le spectro pour détecter la présence de protéines parce que le tryptophane (Trp, W) absorbe la lumière UV avec un pic à 280 nm. La tyrosine (Tyr, Y) le peut aussi, quoiqu'à un degré moindre, et un troisième acide aminé cyclique, la phénylalanine, le peut aussi très faiblement. Tous les peptides pourraient absorber l'UV aux environs de 190-200nm, mais la plupart des tampons absorbent aussi massivement dans cette région qui n'est donc pas très commode pour nous.
Notez que le coefficient d'extinction de chaque protéine variera selon sa richesse en W et Y. Une protéine dépourvue de ces acides aminés passerait devant la cellule optique comme un fantôme, à peine percue.
(B) Par Méthodes colorimétriques.
Ces techniques demandent un traitement de l'échantillon à mesurer par une substance chimique qui, au contact des protéines, produira un changement de couleur que l'on peut quantifier par spectrophotométrie si on dispose d'une courbe standard fiable.
Comme dans le cas de l'absorbance à 280nm, les méthodes colorimétriques réagissent de façon inégales aux différentes protéines. pour une analyse brute, une courbe standard faite avec des quantités connues de BSA convient tout à fait, mais pour des protéines plus pures il vaut mieux s'assurer de disposer d'une courbe standard dont le comportement représente bien celui de notre protéine.
|
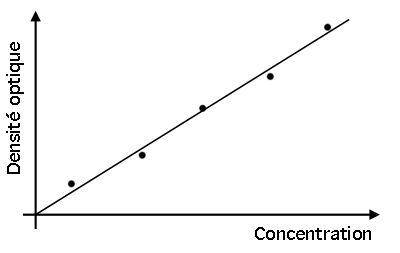
|
Exemple de courbe standard: la mesure de la densité optique (DO) de plusieurs échantillons avec une concentration croissante en protéines donne à chaque fois une DO plus élevée. On peut ainsi tracer une droite qui nous permet de déterminer la concentration en protéines d'un échantillon inconnu grâce à sa propre DO.
|
Toujours avoir plusieurs points sur notre courbe standard: on limite ainsi l'effet de ceux qui auraient eu une mauvaise réaction. C'est comme avec des fonds mutuels, vraiment.
Voici trois méthodes colorimétriques; la méthode de Bradford est probablement la meilleure et la plus utilisée aujourd'hui, mais il en existe plusieurs autres.
|
Méthode du biuret |
Basée sur la réduction du cuivre Cu2+ en Cu+.
Cu+ réagit avec le tryptophane (Trp, W), la tyrosine (Tyr, Y) et la cystéine (Cys, C). Il leur donne une couleur bleue. Le pic d'absorption pour un test du biuret est à 550nm.
Ce test est peu sensible.
Il résiste assez bien aux différents composés présents dans les tampons mais est sensible aux sels d'ammonium, ce qui en fait un mauvais choix pour doser les protéines venant d'être précipitées par ce sel.
|
|
Méthode de Lowry |
Basée sur la réaction du biuret, elle utilise aussi la réduction du Cu2+ en Cu+. Depuis 1951, la méthode de Lowry est la plus citée de toutes dans la littérature scientifique.
Dans la méthode de lowry, le Cu+ est utilisé pour réduire le réactif de Folin (une solutiuon phénolique contenant des composés de tungstène et de molybdène) qui change sa couleur du jaune au bleu. On lit la réaction à 750 nm.
Plus sensible que la réaction du biuret, la méthode de Lowry est utilisée pour des quantités de 2 — 100 ug.
Elle est sensible à plusieurs agents, dont l'EDTA, le DTT, le b-mercapto, l'Hepes, le Tris, le triton X-100, le NP-40, etc.
|
|
Méthode de Bradford |
|

|
Le bleu de Coomassie se lie à l'arginine (Arg, R), la tyrosine (Tyr, Y), le tryptophane (Trp, W), l'histidine (His, H) et la phénylalanine (Phe, F) (surtout à R; huit fois plus qu'aux autres en fait).
Il est assez insensible aux agents des tampons, mais est sensible aux détergents.
En solution, il a une forme cationique rouge qui absorbe à 470nm. Lié aux protéines, il a une forme anionique bleue qui absorbe à 595nm.
Parce que les deux spectres d'absorption se chevauchent un peu, une courbe standard de Bradford n'est pas parfaitement linéaire sur toute sa distance (quoiqu'en disent les commerçants!), un point souligné par Brafdord lui-même dans son papier original. Elle l'est quand même suffisament pour nos besoins.
La méthode de Bradford est encore plus sensible que celle de Lowry (0,2 — 20 ug de protéines).
|
|
5.3.2.2 Présence de protéines particulières
L'absorption aux UV et les méthodes colorimétriques c'est bien, surtout si on veut juste savoir par où passent les protéines durant la purification. Il vient cependant un temps où, confiants de ne pas avoir flushé nos protéines aux égoûts, on veut savoir si parmi elles se trouvent la protéine qui nous intéresse.
Un critère utile de notre protéine pour la reconnaître parmi les autres est sa taille; un autre tel critère est sa charge.
5.3.2.2.1 Gel de SDS
Le système le plus utilisé pour l'analyse de la taille des protéines d'un mélange est le gel de SDS (ou SDS-PAGE, pour sodium docecyl sulfate -polyacrylamide gel electrophoresis).
La séparation électrophorétique d'une protéine, tout comme celle de n'importe quelle molécule chargée d'ailleurs, est l'effet de son mouvement sur ou dans une matrice appropriée soumise à un courant électrique. Plus une molécule est massive ou biscornue, plus elle se déplacera lentement; plus elle sera petite, plus elle ira vite.
Il serait avantageux, comme ce l'est pour l'ADN, de pouvoir séparer les protéines les unes des autres par une simple séparation électrophorétique ne dépendant que de la taille des molécules. Mais alors que l'ADN est uniformément chargé et que sa structure est essentiellement la même quand il est sous forme linéaire, les protéines, elles, varient énormément non seulement en poids mais aussi en charge et en forme. Pour les séparer selon leur masse uniquement, il faut contrecarrer l'effet de leur charge et de leur forme.
Ce tour de passe-passe est accompli en mettant 1% de SDS dans le gel d'électrophorèse et dans le tampon de migration. On utilise aussi 0,1M β-mercaptoethanol (HO-CH2-CH2-SH) dans le tampon de largage des échantillons, et ceux-ci sont bouillis de surcroît avant la migration sur gel.
Le sodium dodecyl sulfate (SDS) est un détergent anionique qui dénature les protéines en enveloppant la chaîne polypeptidique selon un ratio de masse de 1,4 pour 1. Le SDS confère aux protéines une charge négative proportionnelle à leur longueur; plus elles sont longues, plus il y a de SDS. Les polypeptides deviennent donc des chapelets de charges négatives partageant une même densité de charge par unité de longueur.
Le β-mercaptoethanol, lui, (un agent réducteur qui ne sent pas bon), en combinaison avec la chaleur, brise les ponts disulfures internes des protéines que la chaleur ont dénaturées. Il brise aussi les éventuels ponts disulfures inter-protéiques.
Les protéines ainsi dépliées et uniformément chargées en fonction de leur masse peuvent dès lors être séparées par électrophorèse, de la cathode (-) à l'anode (+). Puisqu'elles sont uniformément chargées par le SDS, leur vitesse de migration sur gel sera proportionnelle à leur seule masse, les petites protéines étant les plus rapides.
On utilise d'ordinaire un gel discontinu: c'est à dire qu'en haut du gel de séparation proprement dit, (le "running gel;" en bon franglais) se trouve une bande plus petite d'acrylamide de composition différente, le gel de concentration ou stacking gel.
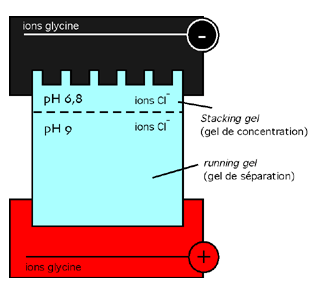
|
Alors que le gel de séparation a des pores petits et un pH de 9, le stacking gel a des pores plus grands et un pH de 6,8. La taille des pores dans un gel de polyacrylamide est fonction du degré de pontage entre les chaînes d'acrylamide, pontage qui est proportionnel à la quantité de N,N,N',N',-méthylène bisacrylamide dans le gel; plus il y en a, plus les chaînes sont pontées et plus petits sont les pores.
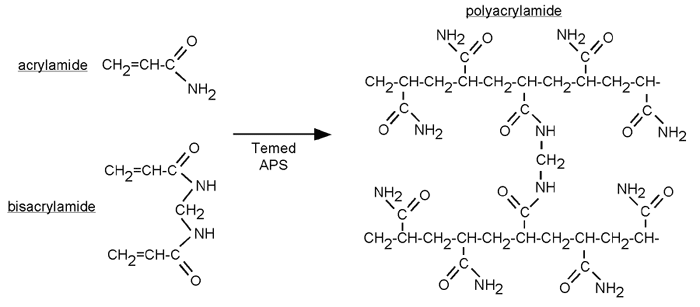
|
De plus, les ions des deux gels au moment du dépôt des protéines sont des ions chlorure alors que ceux du tampon sont des ions glycine. Les ions glycine qui entrent dans le gel de concentration à pH 6,8 sont près de leur point isolélectrique et ont une très faible mobilité. Les ions chlorure, eux, conservent une grande mobilité. Les protéines chargées par le SDS sont plus lentes que le chlorure mais plus rapides que la glycine.
|
Quand le courant est appliqué, les ions chlorures filent vers l'anode, laissant derrière elles un vide ionique temporaire qui n'a qu'une faible conductivité.
Manquant d'électrolytes dans leur entourage immédiat, les protéines dans le gel de concentration ne peuvent pas migrer vers l'anode. En fait, elles ne peuvent bouger que quand le front d'ions glycine finit par les rattraper, parce que ces ions remplacent alors les ions chlorure comme électrolytes. Même alors, parce que les grands pores du gel de concentration permettent aux protéines d'être aussi rapides que les ions glycine, elles ne se séparent pas les unes des autres mais sont toutes ramassées au passage par le front d'ions.
(Imaginez que les protéines sont des barques échouées sur la grève, nez pointé vers la terre ferme, et qu'elles sont soudain poussées par une vague (le front d'ions). Vous aurez une idée assez correcte de ce qui se passe dans cette partie du gel. Même les barques les plus rapides ne peuvent pas aller plus vite que la vague, parce qu'elles manqueraient tout simplement d'eau pour les porter).
Ce front d'ions qui pousse et empile les protéines devant lui induit la concentration de toutes les protéines en une mince bande qui migre avec le front jusqu'au bout du gel de concentration.
Quand le front d'ions atteint la discontinuité de pH et de porosité entre le gel de concentration et le gel de séparation, le pH soudain plus élevé permet aux ions glycine de prendre leur envol. Ils deviennent beaucoup plus chargés et rapides et dépassent les protéines.
|
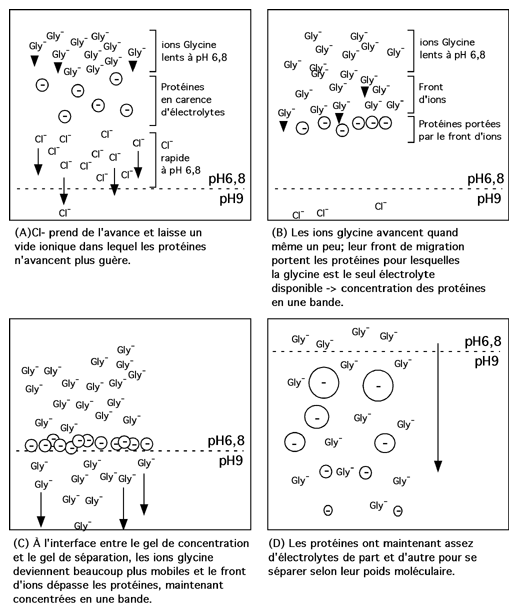
|
Maintenant qu'il y a assez d'électrolytes en avant d'elles, les protéines peuvent migrer librement en fonction de leur masse. (La porosité du gel, qui est plus faible dans le gel de séparation que dans le gel de concentration, assure que les ions glycine seront plus rapides que les protéines).
Soyons honnêtes avant de continuer: il nous faut avouer que la mobilité des protéines dans un gel de SDS n'est pas absolument proportionnelle à leur masse. En effet, on peut observe une variation de vitesse de migration par rapport à la masse dans une majorité de protéines, allant de très petites variations jusqu'à des résultats franchement désarçonnants. La cause peut en être variable: par exemple, certaines protéines ont naturellement une faible affinité pour le SDS et le ratio masse/charge s'y trouve modifié. Certaines modifications post-traductionnelles peuvent aussi jouer un rôle: la glycosylation interfère avec la liaison au SDS. Il convient donc de ne pas se désespérer si nos protéines purifiées ont sur gel de SDS une taille apparente différente de ce à quoi on s'attendait!
Les très petites protéines (moins de 15 kDa), en outre, courent de façon très aléatoires dans les gels de glycine. C'est qu'elles peuvent être capturées dans des micelles de SDS qui se tiennent près du front de migration. On peut les en libérer en utilisant la tricine à la place de la glycine dans le tampon de migration. La tricine aide à la libération de ces petites protéines. On offre commercialement des gels de SDS Tris-tricine pour l'étude des protéines de plus petit poids moléculaire.
Une fois les protéines dûment séparées selon leur taille, il reste à révéler leur présence. Les deux approches les plus courantes sont de les colorer, ou d'utiliser un anticorps spécifique qui permette de localiser une réaction chimique quelconque (mais donnant un résultat visible) là où se trouve une protéine particulière (voir à ce sujet la technique de l'immunobuvardage de type western, un peu plus loin).
On peut colorer les protéines directement sur le gel, ou les colorer après les avoir transféré sur une membrane (quoique dans ce dernier cas, il ne s'agisse d'ordinaire que d'un rapide contrôle pour vérifier la qualité du transfert; on colore presque toujours les protéines directement dans le gel).
Les techniques les plus courantes sont la coloration au bleu de Coomassie et au nitrate d'argent. Le bleu de Coomassie servait jadis à colorer les chandails de laine en bleu; son nom commémore la prise de la ville Ashantie de Kumasie, au Ghana, pendant l'époque coloniale. Le bleu de Coomassie colore les protéines de façon à peu près égale mais est de dix à cent fois moins sensible que la technique de coloration au nitrate d'argent.
Cette dernière repose sur la réduction, facilitée par la présence des protéines, de sels d'argent en argent métallique (qui précipite). Il existe plusieurs protocoles de coloration au nitrate d'argent (choisissez la vôtre en fonction de sa facilité d'exécution et du temps qu'elle requiert)! Cette technique est cependant sensible à plusieurs facteurs et ne colore pas toutes les protéines de façon égale.
5.3.2.2.2 Gel non-dénaturant "Blue native".
Une variante du gel SDS permet de greffer une charge aux protéines ou aux complexes protéiques sans les dénaturer. À la place du SDS, un détergent, on utilise le bleu de Coomassie qui donnera une charge négative aux protéines en se collant (principalement) sur ses résidus arginine. La distrubution des charges ne permet pas de définir un ratio charge/masse aussi constant qu'avec le SDS, mais au moins les protéines natives migrent dans le même sens. Cette technique est appelée "blue native electrophoresis". (Schagger H, von Jagow G. Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal Biochem. 1991 Dec;199(2):223-31).
benoit.leblanc@usherbrooke.ca