Chapitre 1:
Question 1.1: L'histidine, la lysine et l'arginine sont trois acides aminés basiques. Le pK de leur chaîne latérale est respectivement d'environ 6, 10,5 et 12,5. Lesquels de ces acides aminés sont les plus basiques? Et quel serait leur caractère à un pH à peu près neutre?
Réponse: Le pK décrivant l'équilibre NH3+ <=> NH2 est de 6 pour l'histidine. C'est donc dire qu'aux environs de pH 6, les deux formes sont en quantités équivalentes. Pour la lysine et l'arginine, ce pK est de 10,5 et de 12,5. Cela signifie que la chaîne latérale de ces acides aminés reste sous forme NH3+ sur un bien plus grand spectre de pH; il faut monter le pH au-dessus de 10 et de 12 pour que NH3+ se convertisse en NH2. Ces deux acides aminés ont donc un caractère basique plus important que l'histidine.
À pH à peu près neutre, l'histidine aurait un faible caractère basique, car l'équilibre NH3+ <=> NH2 serait déjà un peu déséquilibré vers la droite. Les deux autres acides aminés, par contre, sont résolument sous forme NH3+ et ont donc un fort caractère basique.
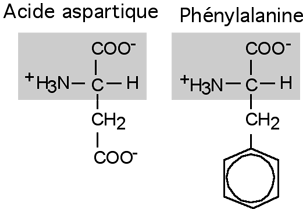
Question 1.2: L’aspartame, cet édulcorant bien connu, est un simple dipeptide: l’aspartylphénylalanine méthyl ester. C’est le groupement carboxylique libre de la phénylalanine qui est estérifiée en C-O-CH3. Dessinez la structure de l’aspartame (vous pouvez aller voir à quoi ressemblent la phénylalanine et l'acide aspartique si vous voulez). Ionisez les groupes comme si le pH était à 7. Quels seraient les groupes à considérer pour estimer le pI de l’aspartame?
Réponse: Voyons d’abord ce que nous dit la question. L’aspartame est un dipeptide qui contient de l’aspartate et de la phénylalanine. Voici la structure de ces deux acides aminés:
On nous dit aussi que le groupe carboxylique de la phénylalanine est estérifié par par un goupe méthyl (un groupement ester forme un lien
-C - O - C- ):
||
O
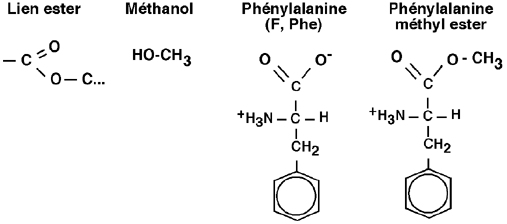
Puisque l'extrémité carboxylique de la phénylalanine est occupée, le lien peptidique du dipeptide ne peut donc se former qu’entre le groupe carboxylique de l’acide aspartique et le groupe aminé de la phénylalanine methyl ester: (Vous pouvez orienter les groupes latéraux comme bon vous semble).

Point de vue pI maintenant: Le pI est le pH auquel les charges positives et négatives d’un peptide s’annulent. Dans le cas qui nous occupe, nous avions au départ cinq groupement chimiques ionisables:
- l’extrémité aminée de l’acide aspartique NH2 < - > NH3+, pK= 9,6
- l’extrémité aminée de la phénylalanine NH2 < - > NH3+, pK= 9,13
- l’extrémité carboxylique de l’acide aspartique COO- < - > COOH, pK=2,19
- l’extrémité carboxylique de la phénylalanine COO- < - > COOH, pK=1,83
- et finalement la chaîne latérale de l’acide aspartique COO- < - > COOH, pK= 3,65
Le groupe carboxylique de la phénylalanine fait maintenant partie d’un lien ester C-O-C; il n’est donc plus ionisable. Le groupe carboxylique de l’acide aspartique et le groupe aminé de la phénylalanine sont maintenant unis par un lien peptidique; ils ne sont plus ionisables ni l’un ni l’autre.
Il ne reste donc que deux groupes ionisables: le groupe aminé de l’acide aspartique ainsi que le groupe carboxylique de sa chaîne latérale. Le pI de l’aspartame se calculera en trouvant le pH où l’ionisation de l’un compensera celle de l’autre.
Question 1.3: La proline est un acide iminé plutôt qu'aminé. Quelle est sa caractéristique principale donnant de l'importance à cette distinction?
Contrairement aux autres acides aminés, la proline n'a pas une chaîne latérale "pendant dans le vide", si vous me passez l'expression. Sa chaîne latérale se recourbe et vient former un lien avec le N du groupe aminé. Cet aspect aura une importance majeure sur l'influence de la proline sur les structures secondaires.
Question 1.4: Lequel de ces peptides est le plus acide? Le plus hydrophobe?
(a) MRHDCEILDFEWDEAGGEGDSETD
(b) MHGFHIKLKDKQLRRKKIPREWDKL
(c) FWYKPYWFVNAAVFWYALCDFYR
(d) AQNDFMLKCSEFKDYTREDCAQRD
(a) contient le plus de résidus acides (D et E); (c) contient le plus de résidus hydrophobes (A, V, L, I, P, W, F, Y).
Question 1.5: Certains organismes utilisent certains codons d'une manière différente. Quelles sont les limitations imposées à ce phénomène?
Les protéines que l'évolution a sélectionné ne peuvent pas supporter trop de mutations sans devenir non-fonctionnelles. Le changement d"identité d'un codon phénylalanine pour un codon tryptophane partout dans le génome causerait des dommages irréparables. L'utilisation alternative de codons est donc limitée à ce qui ne causera pas trop de dégâts. Des codons STOP peuvent dans certains cas coder pour un acide aminé sans causer de catastrophe, puisque cette modification ne change rien à la séquence des protéines en général. certaines organelles ayant leur propre génome, comme les mitochondries, peuvent utiliser des codons d'une façon différente du noyau. Il est alors peu probable que des gènes puissent facilement passer de l'un à l'autre, puisque ces gènes ne coderaient pas pour la même séquence protéique dans l'un et l'autre cas!
Question 1.6: Petit exercice sur l'internet. Rendez-vous sur le site du NCBI et cherchez les séquences en acides aminés des récepteurs nucléaires RXR alpha1, RXR beta1, et RXR gamma2 de la souris (mus musculus). Sauvez ces séquences quelque part en format texte et rendez-vous ensuite sur le site de l'institut européen de bioinformatique. Vous y trouverez une liste de plusieurs outils bioinformatiques. Choisissez le programme ClustalW qui sert à faire des alignements. (ClustalW est disponible ailleurs, aussi ne vous gênez pas si vous préférez un autre site). Pour comparer les trois séquences, copiez-collez chacune d'entre elles dans la fenêtre à cet effet, l'une à la suite de l'autre, en les séparant par le symbole > et le nom de la protéine, suivie par un point. En comparant l'alignement des trois séquences, où remarque-t-on le plus de disparité entre elles?
RXRalpha1. ---MDTKHFLP-------------------------------LDFSTQVNSSSLN----- 21
RXR beta1. MSWATRPPFLPPRHAAGQCGPVGVRKEMHCGVASRWRRRRPWLDPAAAAAAAGEQQALEP 60
RXRgamma2. ------------------------------------------------------------
RXRalpha1. ----------SPTGRGSMAVPSLHPS-LGPGIG--SPLGSPGQLHSPISTLSSPINGMGP 68
RXR beta1. EPGEAGRDGMGDSGRDSRSPDSSSPNPLSQGIRPSSPPGPPLTPSAPPPPMPPPP--LGS 118
RXRgamma2. ---------MATNKERLFAPGALGPGSGYPGAG--FPFAFPGALRGS------------P 37
. . . : : *. * * . * .. .
RXRalpha1. PFSVISSPMGPHSMSVPTTPTLGFGTGSPQLNSPMNP----VSSTEDIKPPLGLNGVLKV 124
RXR beta1. PFPVISSSMGSPGLPPPAPPGFSGPVSSPQINSTVSLPGGGSGPPEDVKPPVLGVRGLHC 178
RXRgamma2. PFEMLSPSFRGLGQPDLPKEMASLSVETQSTSS-----------------------EEMV 74
** ::*..: . . . . . : . .*
RXRalpha1. PAHPSGNMASFTKHICAICGDRSSGKHYGVYSCEGCKGFFKRTVRKDLTYTCRDNKDCLI 184
RXR beta1. PPPPGGPGAG--KRLCAICGDRSSGKHYGVYSCEGCKGFFKRTIRKDLTYSCRDNKDCTV 236
RXRgamma2. PSSPSPPPPPRVYKPCFVCNDKSSGYHYGVSSCEGCKGFFRRSIQKNMVYTCHRDKNCII 134
*. *. . : * :*.*:*** **** *********:*:::*::.*:*: :*:* :
RXRalpha1. DKRQRNRCQYCRYQKCLAMGMKREAVQEERQRGKDRNENEVESTSSANEDMPVEKILEAE 244
RXR beta1. DKRQRNRCQYCRYQKCLATGMKREAVQEERQRGKDK-DGDGDGAGGAPEEMPVDRILEAE 295
RXRgamma2. NKVTRNRCQYCRLQKCFEVGMSKEAVRNDRNKKKKEVKEEGSPDSYELSPQLEELITKVS 194
:* ******** ***: **.:***:::*:: *.. . : . . . : * :..
RXRalpha1. LAVEPKTETYVEAN--MGLNPSSPNDPVTN-----ICQAADKQLFTLVEWAKRIPHFSEL 297
RXR beta1. LAVEQKSDQGVEGPGATGGGGSSPNDPVTN-----ICQAADKQLFTLVEWAKRIPHFSSL 350
RXRgamma2. KAHQETFPSLCQLGKYTTNSSADHRVQLDLGLWDKFSELATKCIIKIVEFAKRLPGFTGL 254
* : . : . :. . : :.: * * ::.:**:***:* *: *
RXRalpha1. PLDDQVILLRAGWNELLIASFSHRSIAVKDGILLATGLHVHRNSAHSAGVGAIFDRVLTE 357
RXR beta1. PLDDQVILLRAGWNELLIASFSHRSIDVRDGILLATGLHVHRNSAHSAGVGAIFDRVLTE 410
RXRgamma2. SIADQITLLKAACLDILMLRICTRYTPEQDTMTFSDGLTLNRTQMHNAGFGPLTD-LVFA 313
.: **: **:*. ::*: :. * :* : :: ** ::*.. *.**.*.: * ::
RXRalpha1. LVSKMRDMQMDKTELGCLRAIVLFNPDSKGLSNPAEVEALREKVYASLEAYCKHKYPEQP 417
RXR beta1. LVSKMRDMRMDKTELGCLRAIILFNPDAKGLSNPGEVEILREKVYASLETYCKQKYPEQQ 470
RXRgamma2. FAGQLLPLEMDDTETGLLSAICLICGDRMDLEEPEKVDKLQEPLLEALRLYARRRDPAKP 373
:..:: :.**.** * * ** *: * .*.:* :*: *:* : :*. *.::: * :
RXRalpha1. GRFAKLLLRLPALRSIGLKCLEHLFFFKLIGDTPIDTFLMEMLEAPHQAT---------- 467
RXR beta1. GRFAKLLLRLPALRSIGLKCLEHLFFFKLIGDTPIDTFLMEMLEAPHQLA---------- 520
RXRgamma2. YMFPRMLMKITDLRGISTKGAERAITLKMEIPGPMPPLIREMLENPEMFEDDSSKPGPHP 433
*.::*:::. **.*. * *: : :*: *: .:: **** *.
RXRalpha1. -------------------------
RXR beta1. -------------------------
RXRgamma2. KASSEDEAPGGQGKRGQSPQPDQGP 458
Le N-terminus des trois protéines est très différent. La partie centrale est très homologue, et la partie C-terminale est similaire chez alpha et beta mais dissemblable chez gamma.
Chapitre 5
Question 5.1 Un polypeptide digéré par une protéase libère les peptides suivants. Ils sont purifiés sur une colonne échangeuse de cations. Donnez l’ordre de leur élution à pH 7 en utilisant un gradient de KCl de 100mM à 1M. Justifiez votre choix.
EEEDFPPGPHRQKKK
GGILPA
TSGKAY
GHACGF
DDEDEAA
VNRKGW.
La colonne est échangeuse de cations, c’est donc dire qu’elle est chargée négativement et qu’elle aura tendance à retenir les charges positives. Les résidus avec une chaîne latérale basique (H, R, K) seront donc retenus davantage que les autres. Comptons les charges positives de chacun des peptides:
EEEDFPPGPHRQKKK = 1 H, 1 R, 3K
GGILPA = rien
TSGKAY = 1 K
GHACGF = 1 H
DDEDEAA = rien
VNRKGW = 1 R, 1 K
GGILPA et DDEDEAA sortiront en premier parce qu’ils ne seront pas retenus par la résine. Ils ne contiennent pas d’acides aminés basiques. Ensuite, entre les deux peptides avec un résidu basique chacun, c’est GHACGF qui sortira le premier parce que son histidine est moins basique (pk = 6) que la lysine de TSGKAY ( pK= 10,53). VNRKGW sortira ensuite, avec ses deux charges positives et EEEDFPPGPHRQKKK sortira le dernier parce qu’il a 5 résidus basiques, même s’il en compte aussi 4 acides.
Question 5.2 Une purification par gradient de sucrose donne 12 fractions séparés en partant du haut du tube. Sur gel SDS, voici ce qu’elles donnent comme patron. Que signifie le patron de la piste 7?
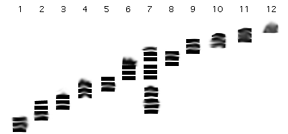
Les protéines se sont séparées en fonction de leur densité, qui va souvent de pair avec leur poids moléculaire. La piste 7 présente une anomalie en ce qu’on y retrouve plusieurs protéines de poids très différents. On peut supposer que plusieurs de ces protéines étaient associées en un ou plusieurs complexes dont la densité globale était différente des densités individuelles de ses composantes.
Question 5.3 Vous purifiez sur colonne d’échange d’ions, avec un tamnpon Tris à pH 7, un polypeptide de séquence
GGLDNREDSARTILEKKERIEDQTHTSGPQMRHNVMRHNKLILDED.
Quel moyen de détection recommandez-vous pour suivre l’élution du peptide? Justifiez votre réponse.
Comme le peptide ne contient pas de tryptophane ou même de tyrosine, on ne peut pas utiliser la détection par spectrophotométrie à 280nm. Dans un tampon tris, la lecture à 200nm serait également impossible.
Du côté colorimétrique, on évitera les méthodes basées sur la réaction du Biuret, qui repose aussi sur la présence de W, Y ou C. (Il n’y a pas de C non plus dans le peptide!)
On pourrait utiliser la méthode de Bradford.
Question 5.4 Vous disposez d'un extrait cellulaire total duquel vous voulez extraire une glycoprotéine encore mal connue. Décrivez cinq techniques utilisées pour la purifier, si vous savez déjà qu'elle a un pI de 9, que sa séquence interne contient la séquence RRKGHHHHHHSM et qu'elle se lie à la protéine SP1. Justifiez vos choix et soulignez le ou les avantages de chacune des techniques que vous choisissez.
exemple de réponse acceptable:
1- Glycoprotéine: colonne d’affinité pour les protéines glycosylées, comme une WGA (wheat germ agglutinin) ou ConA (concanavalin A).
2- La protéine est très basique (pI de 9): chromatographie sur échangeuse de cations à pH 7-8.
3- Séquence interne contient HHHHHH: chromatographie d’affinité pour le métal immobilisé (IMAC), comme le Ni-NTA.
4- Elle se lie à SP1: chromatographie d’affinité avec une résine couplée à du SP1 recombinant.
5- Et finalement, on peut toujours enrichir une protéine par filtration sur gel.
Question 5.5 Vous purifiez un enzyme qui contient quatre sous-unités de 65 kD.
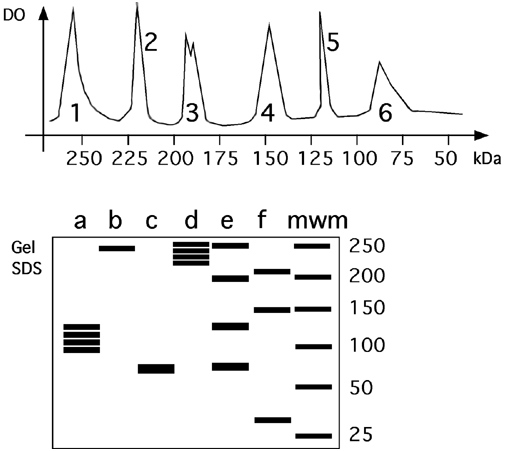
(a) Dans des conditions non-dénaturantes, dans quel pic d’élution d’une colonne de filtration sur gel (1 à 6, ci-contre) vous attendez-vous à retrouver cet enzyme?
(b) Sur gel SDS, quelle piste contiendrait l’enzyme?
(c) Et si on pontait les protéines au dimethylsuberimidaqte avant la purification?
réponse:
(a) Pic 1, qui pourrait correspondre à un tétramère de sous-unités de 65 kD (pour 260 kD).
(b) Le SDS va briser le tétramère en ses sous-unités de 65 kD: C’est probablement la piste C qui est la bonne, quoiqu’on ne puisse pas absolument exclure que le pic 1 ait aussi contenu des complexes contenant des sous-unités de 60 Kd et de poids moléculaires supérieurs comme on en retrouve dans la piste e.
(c) Si on pontait les sous-unités au suyberimidate, le patron ressemblerait à la piste e, qui contient des bandes correspondant à différentes combinaisons des sous-unités du complexe (65 + 130 + 195 + 260)