| Retour à la page d'accueil | Section précédente | Section suivante |
7.2.1 Sensibilité à la nucléase micrococcale
La nucléase micrococcale a beaucoup de difficulté à couper l'ADN complexé à un nucléosome. On peut donc utiliser cet enzyme pour couper la chromatine entre les nucléosomes, et juger ainsi de l'état de la chromatine.
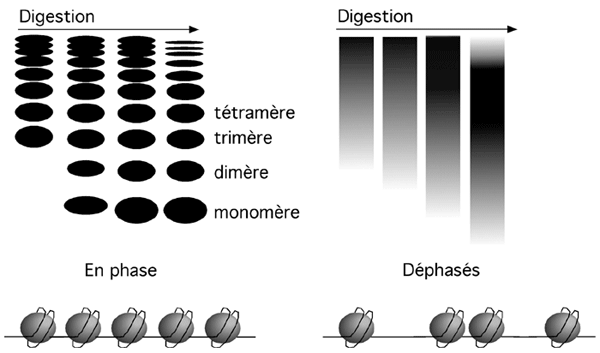
L'ADN d'une cellule coupé à la nucléase micrococcale et séparé sur gel d'agarose ressemble à une échelle de fragments dont la taille est un multiple d'environ 147pb. Ils correspondent à des monomères, dimères, trimères, etc. de nucléosomes. Quand on tranfère cette échelle sur une membrane et qu’on l’hybride avec une sonde d’ADN, on observera une échelle si la région où s’hybride la sonde a des nucléosomes en phase (régulièrement déposés) ou encore une traînée si les nucléosomes sont déphasés (déposés irrégulièrement).
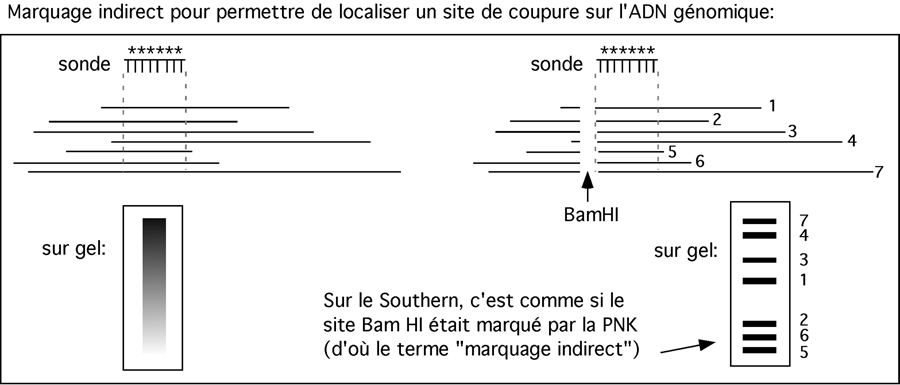
Pour juger d'un site spécifique de coupure par la nucléase micrococcale, on peut effectuer un marquage indirect ("indirect end-labelling"). Pour ce faire, après traitement à la nucléase micrococcale, l'ADN est coupé avec une endonucléase qui a un site près de la région qui nous intéresse. On effectue ensuite un transfert de Southern, et comme sonde on en choisit une, pas trop longue, qui hybride juste à l'extrémité du fragment laissé par la digestion à l'endonucléase. Cette hybridation revient au même que d'avoir effectué un marquage au site de restriction, et permet de juger des distances entre ce bout et toute coupure causée par la nucléase micrococcale. (Cette technique fonctionne aussi bien sûr avec toutes les nucléases).
7.2.2 Extension de monomères
Cette technique un peu plus compliquée donne des résultats plus difficiles à analyser mais fournit énormément d'information sur la position des nucléosomes.
On commence par digérer de la chromatine par la nucléase micrococcale jusqu'à l'obtention de nucléosomes monomériques (les conditions de digestion sont à déterminer empiriquement et préalablement).
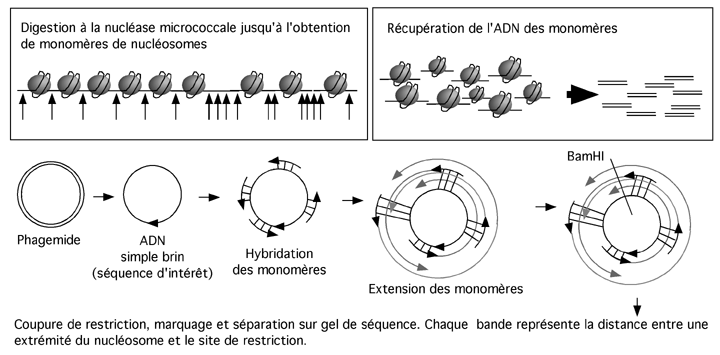
Les monomères sont ensuite purifiés pour en récupérer l'ADN.
On aura au préalable préparé un phagemide de même séquence que la région que nous voulons étudier dans la chromatine. Ce phagemide servira à faire de l'ADN simple brin (on doit préparer les deux brins indépendamment pour déterminer la position de chacune des deux extrémités des monomères).
L'ADN récupéré des monomères de nucléosome est dénaturé et hybridé sur l'ADN simple brin du phagemide; il est ensuite allongé par la polymérase de Klenow. Chaque monomère donnera naissance à un fragment d'ADN dont la synthèse commence à une position qui lui est propre.
L'ADN obtenu après l'extension par la Klenow est ensuite coupé avec une endonucléase coupant une fois dans le plasmide, ce qui génère une échelle de bandes: chaque bande de cette échelle a une taille correspondant à la distance entre l'extrémité distale du monomère et le site de restriction. Cela nous donne la positon d'une des extrémités de chaque nucléosome.
On répète l'opération avec l'autre brin pour obtenir la position de l'autre extrémité.
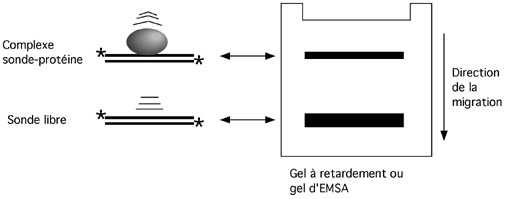
Cette technique, la plus simple qui soit pour étudier une interaction entre un acide nucléique et des protéines, repose sur le fait qu’un complexe ADN- protéine ou ARN-protéine migrera moins vite dans un gel non-dénaturant qu’un ADN ou un ARN nu. Ce retard de migration permet de juger au premier coup d’oeil si une séquence particulière d’ADN ou d’ARN a été reconnue et liée par une protéine. On l’appelle aussi gel-shift ou gel à retardement, ce qui le fait sonner comme une bombe.
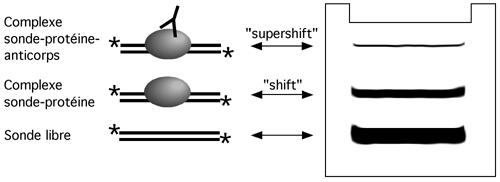
On peut vérifier la spécificité de l'interaction ADN-protéine par l'incubation préalable du complexe avec un anticorps qui se fixe à la protéine. Le complexe ternaire ADN-protéine-anticorps a une migration encore plus retardée, qu'on qualifie de supershift. Notez qu'il arrive qu'un anticorps qui fonctionne très bien en western ne vale pas grand chose en supershift. Peut-être que son épitope est moins accessible, ou que son affinité n'est pas assez forte pour résister aux conditions présentes pendant la migration. On peut aussi trouver des anticorps qui interfèrenet avec la liaison ADN-protéine; ceux-là, au lieu de provoquer un supershift, vont même inhiber le gel-shift ordinaire.
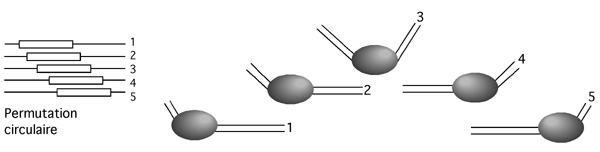
Certaines protéines font se courber l’ADN. Dépendant de l’endroit où elle se lie sur une sonde d’ADN, une protéine qui fait courber l’ADN affectera de façon différente la migration sur le gel d’EMSA. Plus la courbure est au centre de la sonde et plus la migration est retardée. On peut utiliser une même sonde en y déplaçant le site de reconnaissance de la protéine pour juger de l’habileté d’une protéine à courber l’ADN.
7.2.4 SELEX (Systematic Evolution of Ligands by EXponential enrichment)
Le SELEX est une approche de sélection progressive des séquences d'ADN interagissant avec une ou plusieurs protéines. Son algorithme de base ressemble à ceci:
On part d'un ensemble d'oligonucléotides dont la séquence est aléatoire; cela veut dire qu'on y trouve toutes les séquences possibles -incluant les séquences reconnues par notre protéine, ainsi qu'une énorme majorité de séquences qu'elle ne reconnaît pas. Ces oligonucléotides contiennent des séquences permettant leur amplification par PCR: par exemple, ils pourraient avoir une séquence comme ACGTCCGTGCNNNNNNNNNNNNNNNNNNCCGTCGTGCA. Une ronde d'amplification par PCR avec des amorces ACGTCCGTGC et TGCACGACGG les amplifierait tous, quelque soit la séquences de la région NNNNNNNNNNNNNNNNNN.
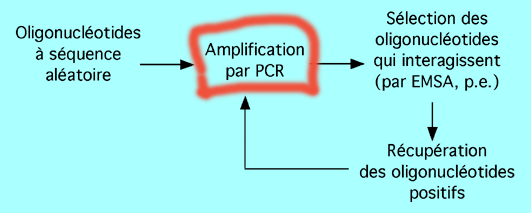
Ce "pool" de séquence est ensuite soumis à une sélection. In vitro, le plus simple est de faire un EMSA avec le pool en entier; les rares séquences qui sont reconnues par la protéine sont retardée lors d'une migration sur gel et le reste est éliminé. Les bandes retardées sont récupérées, et comme elles ne contiennent que peu de matériel, elles sont à nouveau amplifiées par PCR. Comme il est possible que des contaminants se soient glissées à travers les mailles du filet, un nouvel EMSA est pratiqué sur ces séquences réamplifiées; les bandes retardées sont une fois de plus purifiées. On peut exécuter ce cycle plusieurs fois. Les séquences sont à la fin séquencées et on peut alors déterminer celles pour lesquelles la protéine a le plus d'affinité.
Cette technique peut aussi se faire à partir de matériel cellulaire. On commence par ponter les protéines et l'ADN avec du formaldéhyde à même la cellule. Ensuite, on morcelle la chromatine (par sonication, par exemple). Les fragments récupérés sont immunoprécipités avec un anticorps contre la protéine; le pontage est renversé par traitement à la chaleur et l'ADN récupéré. Les fragments d'ADN sont alors rendus francs par traitement à la Klenow et on leur ajoute des adapteurs par ligation pour permettre une amplification par PCR. Aprèes amplification, on peut soit utiliser un EMSA comme ci-haut ou recommencer une ronde d'immunoprécipitation, au choix de l'expérimentateur.
L’exonucléase III a une activité 3’-5’ exonucléase, mais ne s’attaque qu’aux extrémités 5’ proéminentes ou franches. Elle ne coupe pas l’ADN simple-brin, ni à partir d’extrémités 3’ proéminentes.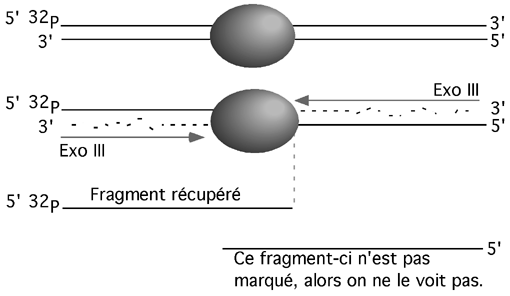
Cette activité de l’exo III est utile pour déterminer les limites d’une interaction entre une protéine et un fragment d’ADN. Si on marque l’extrémité 5’ du fragment au 32P, et que l’on traite celui-ci à l’exonucléase III pendant qu’une protéine en protège une partie, l’exonucléase III ne pourra digérer l’ADN que jusqu’à ce qu’elle entre en contact avec cette protéine, qui lui barre le chemin ni plus ni moins. La taille de l’ADN protégé, qu’on détermine sur gel dénaturant, permet alors de déterminer jusqu’où s’étendait l’interaction ADN-protéine.
Un des avantages de l’empreinte à l’exonucléase III est que l’ADN non-protégé par une protéine est entièrement dégradé; une interaction partielle ne génère pas de bruit de fond.
Cette technique, une fois de plus, utilise le fait qu’une protéine déposée sur l’ADN peut le protéger de l’activité de dégradation d’une nucléase. Dans ce cas précis, il s’agit de la DNAse I, une nucléase qui sans couper absolument partout a quand même une affinité générale pour l’ADN et coupe très fréquemment. La DNAse I ne coupe qu’un seul brin à la fois, aussi on marque un seul des brins au 32P pour suivre son comportement sur gel de séquence.
Afin de déterminer précisément la position de cette empreinte, on fait souvent courir à côté des pistes de digestion à la DNAse une réaction de séquence. Quoiqu’on puisse utiliser la technique de Maxam-Gilbert sur la sonde radioactive qui sert aussi pour la réaction d’empreinte, il est plus commode de préparer une échelle de séquence par la réaction de Sanger en utilisant comme amorce un oligonucléotide dont l’extrémité correspond à celle de la sonde marquée (voir figure). Le choix de la technique de Sanger produit beaucoup plus de matériel qui en plus dure plus longtemps (il est marqué au 35S plutôt qu’au 32P, de demi-vie plus courte), utilise un isotope moins dangereux, et permet d’éviter les réactifs très nocifs de la réaction de Maxam-Gilbert (comme le DMS et la piperidine).
La dégradation partielle du fragment d’ADN protégé par la protéine générera une échelle de fragments de tailles différentes. En comparant l’échelle donnée par un ADN nu à celle donnée par un ADN protégé, on remarque que cette dernière présente un trou, ou une empreinte, correspondant aux sites rendus inaccessibles à l’enzyme par la protéine.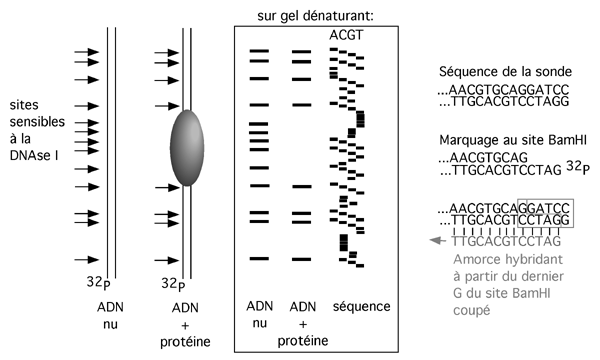
La DNAse I favorise les sites du sillon mineur de l’ADN. Quand l’ADN est en contact avec une surface continue (comme une plaque de verre ou la surface d’un nucléosome, par exemple), elle génère des coupures à toutes les 10 paires de bases, reflétant la fréquence d’hélicité de la spirale d’ADN.
Les empreintes à la DNAse I ont tendance à être grandes et claires.
7.2.7 Empreinte avec d'autres agents
7.2.7.1 Empreinte aux radicaux libres
Le principe de l’empreinte se prête à différents réactifs outre les nucléases. On peut par exemple générer dans le milieu d’incubation des radicaux hydroxyl libres qui attaquent la chaîne de phosphate de l’ADN et cassent un des brins de l’ADN. La réaction pour générer ces radicaux est la suivante:
Fe2+(EDTA4-) + H2O2 -> Fe3+ (EDTA4-) + HO·
C’est le radical HO· qui attaque l’ADN. Il n’a pas de préférence quant à la séquence et tend à couper partout. Une telle empreinte peut donner plus de bruit de fond qu’une empreinte à la DNAse parce que cette dernière, beaucoup plus massive, peut difficilement atteindre des sites situés sous la protéine qui protège l’ADN, alors qu’une petite molécule comme HO· y arrive plus facilement. Pour cette raison, une empreinte générée par les radicaux libres est moins claire qu'une empreinte à la DNAse I.
D’autres radicaux existent pour ce type de réaction, comme par exemple le cuivre-phénanthroline. La décision quant à la technique adoptée revient au chercheur. Votre kilométrage peut varier.
7.2.7.2 Empreinte au DMS Le diméthylsufate, ou DMS, peut méthyler les guanines. Cette réaction de méthylation est plus efficace sur l'ADN nu que sur l'ADN masqué par une protéine et on peut donc l'utiliser pour générer une empreinte. Les méthylguanines sont la cible de la pipéridine chaude, qui coupe un brin de l'ADN à leur niveau (c'est d'ailleurs la réaction utilisée par la méthode de Maxam et Gilbert pour séquencer l'ADN). Bien entendu, comme le DMS n'agit que sur les guanines, l'échelle de digestion que l'on obtient ne contiendra elle aussi que des guanines; on ne pourra pas analyser une région dépourvues de G.
Un avantage du DMS est qu'il peut pénétrer les cellules sans qu'on ait à solubiliser celles-ci au préalable; il se prête donc facilement aux études in vivo. Il est particulièrement actif sur les G se trouvant dans le sillon majeur de l'ADN.
7.2.7.3 Empreinte aux UV ou photofootprinting
Les UVB (280-320 nm) et les UVC (200-230 nm) peuvent induire la formation de dimères de pyrimidines quand deux pyrimidines se voisinent: on obtient alors des CPD, pour cyclobtuane pyrimidine dimers, ou des photoproduits pyrimidine-pyrimidone (6-4PP) . Les 6-4PP se forment à un taux d'environ 15-30% des CPDs. Les 6-4PP peuvent être coupés par la piperidine chaude; les CPD doivent être traités à la T4 endonuclease V et re-traités aux UVA (320-400nm) et à la photolyase.
Cette technique vise plus particulièrement les régions riches en pyrimidines. Elle se prête bien aux techniques in vivo, car aucune perméabilisation membranaire n'y est requise; de plus, l'irradiation peut être très brève si l'intensité de l'irradiation est élevée, permettant de prendre une "photo" de ce qui se passe dans la cellule à un moment précis, avec un minimum de perturbations.
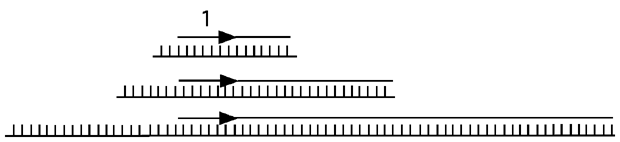
7.2.8 LMPCR et analyses in vivo
Presque toutes les techniques utilisées pour étudier les interactions ADN-protéines in vitro peuvent être utilisées in vivo aussi. Il est juste un peu plus difficile de faire pénétrer les réactifs dans la cellule et d’en récupérer l’ADN par la suite. La technique la plus populaire depuis une dizaine d’années pour récupérer des fragments d’ADN aprèes une réaction d’empreinte in vivo est celle du LMPCR, ou ligation-mediated Polymerase chain reaction. Elle est décrite ci-dessous.
| (1) Récupération de l'ADN génomique. Cet ADN contient des coupures simple-brin, là où la DNAse I ou un autre agent comme le DMS a endommagé la chaîne de groupements phosphates. |
|
| (2) Séparation des brins et annélisation de l'un d'eux avec une amorce 1 spécifique à une région qui nous intéresse. L'autre brin pourra être étudié plus tard lors d'une autre expérience. Notez que les fragments d'ADN dont nous disposons à cette étape sont de tailles différentes, parce que la coupure à la DNAse I n'est que partielle. |
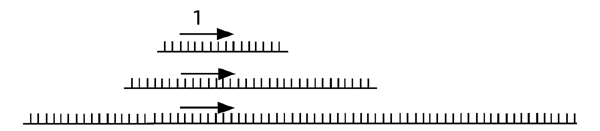
|
| (3) Extension de l'amorce 1. Cette élongation se fait jusqu'au bout de l'ADN qui sert de gabarit, rendant cette extrémité franche ("blunt", comme dirait Coleridge s'il avait fait de la biologie au lieu de la poésie). |
|
| (4) Ligation d'un adapteur double-brin dont une des extrémités est franche et l'autre cohésive. Ces extrémités différentes empêchent l'adapteur de se liguer plusieurs fois. |
|
| (5) PCR entre une deuxième amorce spécifique (l'amorce 2, à côté du site de l'amorce 1) et une amorce (l'amorce 3) semblable à l'extrémité prohéminente de l'adapteur que nous avons ajouté à l'étape précédente. Le premier cycle de ce PCR ne fonctionne que dans un sens, c'est à dire à partir de l,amorce 2, parce que l'amorce 3 n'a pas encore de séquence complémentaire. Cette séquence sera synthétisée lors de ce premier cycle. |
|
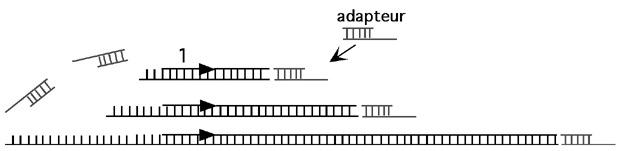
Imaginons qu’on vienne de réaliser une expérience d’empreinte à la DNAse in vivo sur le promoteur du gène Gogosse-1. L’ADN génomique entier de chacune de nos cellules a été coupé par la DNAse I et nous voulons maintenant voir si des sites ont été protégés au niveau de ce promoteur.
Dans la case 1, l’ADN génomique en entier est récupéré. L’ADN qui correspond à notre promoteur est en si petite quantité comparé à tout le reste de l’ADN de la cellule qu’il doit être amplifié par PCR pour pouvoir être visualisé de façon utile.
Dans un premier temps, une amorce spécifique au promoteur qui nous intéresse est utilisée. L’ADN génomique est dénaturé, et l’amorce 1 (ci-dessus) est utilisée pour s’allonger sur tous les fragments correspondant au voisinage du gène d’intérêt. Cette élongation s’arrête quand la polymérase manque d’ADN-gabarit, c’est à dire là où la DNAse I a fait une brèche dans le brin du bas. L’ADN de promoteur ainsi allongé est donc rendu franc à une de ses extrémités.
Dans un deuxième temps, un adapteur (linker) partiellement double-brin est ligué au bout de cette extrémité franche.
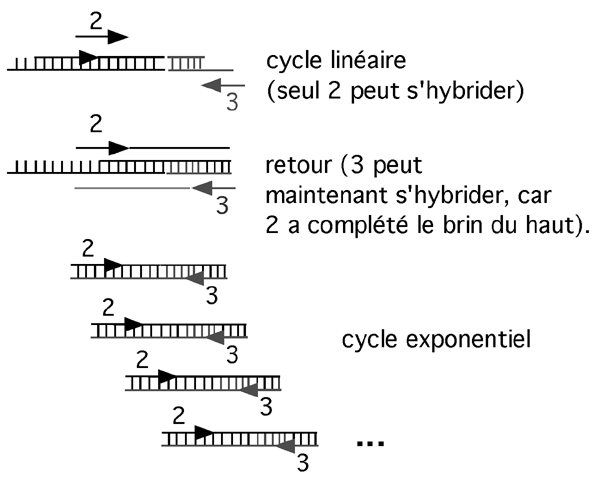
Cet adapteur est franc à un bout et 5’ proéminent à l’autre; cela évite entre autres d’avoir un concatémère d’adapteurs au bout de notre fragment: un seul réussit à s’y liguer. Notez que l’extrémité du fragment d’ADN se trouvant en amont de l’amorce 1 n’a pas pu être allongée et a donc une extrémité proéminente; l’adapteur ne peut pas s’y liguer non plus.
On commence alors la réaction de PCR en utilisant deux nouvelles amorces. La première se trouve un peu en aval de l’amorce 1. (En fait, on pourrait carrément utiliser l’amorce 1 à nouveau, mais des expériences ont démontré que changer d’amorce à cette étape améliore la spécificité des résultats). La deuxième amorce, amorce 3 ci-dessus, correspond à l’extrémité 5’ proéminente de l’adapteur. Comme la séquence sur laquelle cette amorce 3 devrait s’hybrider n’est pas présente sur l’adapteur, le premier cycle de PCR est linéaire: seule l’amorce 2 peut s’hybrider à l’ADN et être allongée.
Comme elle se rend jusqu’au bout du fragment, cette première élongation complète la séquence manquante de l’adapteur; lors du deuxième tour de PCR, l’amorce 3 sera capable de s’hybrider et la réaction sera vraiment exponentielle.
Parmi les traitements disponibles pour effectuer des empreintes in vivo, mentionnons bien sûr...
(a) la DNAse I (il faut alors perméabiliser les cellules pour permettre l’entrée de l’enzyme, et s’assurer de ce qu’il y ait assez de Mg2+ présent dans le milieu);
(b) le diméthylsulfate ou DMS, qui pénètre spontanément dans les cellules et va causer une méthylation des guanines non-protégées, une activité particulièrement efficace dans le sillon majeur de l’ADN; le guanines méthylées peuvent alors être coupées par la piperidine;
(c) et le UV footprinting, ou formation de dimères de pyrimidines par irradiation aux rayons ultraviolets UVB (280-320nm) ou UVC (200-280nm). Ces dimères de pyrimidines peuvent être coupés par une photolyase et la piperidine. Cette empreinte aux UV a l’avantage de ne pas toucher aux cellules du tout, et permet donc d’étudier une cellule très près de son état naturel.
Pour s’assurer de la stabilité d’une interaction ADN-protéine, on peut s’aider un peu en établissant un lien covalent entre les deux. Cela est possible grâce à l’utilisation du fornaldéhyde, HCHO, lequel formera un lien covalent entre protéines ou entre protéines et ADN.
Le gros avantage de ce pontage covalent est qu’il est réversible par la chaleur.
Les expériences d’immunoprécipitation de la chromatine, qui nous ont permis d’en apprendre beaucoup sur les modifications post-traductionnelles des histones, utilisent le pontage au formaldéhyde la plupart du temps.
On peut également ponter les protéines à l’ADN par d’autres techniques. Les rayons UV pourront y parvenir, de même qu’un rayon laser. Dans de tels cas, l’énergie transmise au système permet d’avoir une réaction plus ou moins rapide; un laser assez puissant donnera une image presque instantanée de ce qui colle à l’ADN alors qu’une lampe UV de basse énergie demandera plusieurs minutes, ne révélant que les complexes très stables. (Un problème avec les sources UV de faible énergie est que les rayons UV sont facilement absorbés par le milieu de culture).
7.2.10 SPR (ou surface plasmon resonance), plus communément appelée Biacore
On mesure ici l’interaction entre deux partenaires moléculaires grâce à une machine mesurant l’intensité d’un rayon de lumière. Cette machine est fabriquée par la compagnie Biacore, qui a donnée son nom à l,appareil comme frigidaire l’a fait au réfrigérateur.
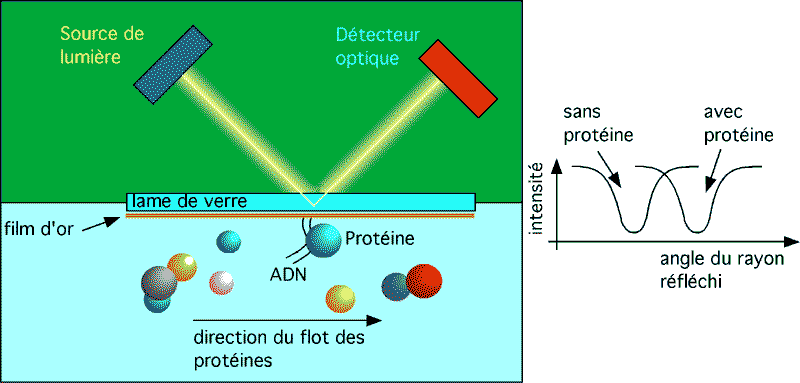
La SPR, ou résonance de plasmon, se produit quand un rayon lumineux est réfléchi sous certaines conditions par un film conducteur à l’interface entre deux milieux d’indices de réfraction différents.
Dans le système Biacore (tm) ces milieux sont (1) la solution dans laquelle se trouvent les molécules à analyser et (2) le verre d’une lame qui sert de senseur. Le film conducteur à l’interface des deux est un très fin film d’or à la surface de la lame de verre.
La résonance de plasmon cause une réduction de l’intensité de la lumière réfléchie à un angle spécifique de réflection. Cet angle varie avec l’indice de réfraction près de la surface du côté opposé à la lumière réfléchie (c’est donc dire du côté de l’échantillon).
Dans le système Biacore (www.biacore.com) , la molécule appât est attachée à la feuille d'or, du côté solution. Les molécules avec lesquelles elle peut interagir sont ajoutées en flot continu. Si l’une d’entre elles interagit avec l’appât, la concentration locale en molcules augmentera dans la région immédiate de l’appât, ce qui fera changer l’indice de réfraction local.
Le changement d’indice de réfraction aura pour effet de changer l’angle pour lequel la lumière réfléchie perd un maximum d’énergie par résonance.
Le système détecte ce un changement et enregistre une interaction. La mesure du niveau d’interaction en fonction du temps permet d’évaluer le taux d’association.
Une unité de résonance (RU) correspond à un changement de 0.0001° dans l’angle donnant une intensité minimale de lumière réfléchie, ce qui pour la plupart des protéines correspond à un changement de concentration de l’ordre de 1 pg/mm2 de surface sur la lame de verre. Le Biacore est donc extrêmement sensible.
7.3 Interactions protéines-ADN in vivo à grande Échelle
7.3.1 ChIP ou Immunoprécipitation de la chromatine
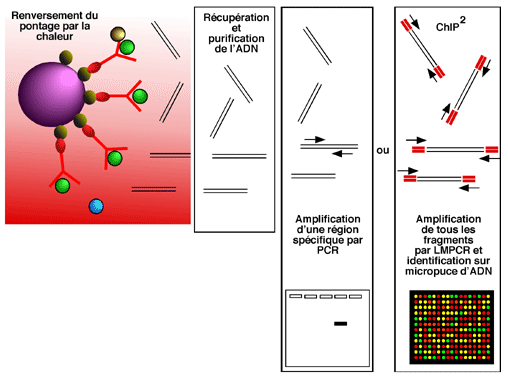
La technique du ChIP, en permettant l'analyse des interactions ADN-protéines à l'échelle de génomes entiers, a profondément marqué la biologie moléculaire depuis quelques années. Elle repose sur la combinaison de différentes techniques, parmi lesquelles l'immunoprécipitation, le pontage covalent, et le PCR.
Dans sa forme la plus simple, le ChIP permet de vérifier la présence d'une protéine donnée à un endroit donné dans le génome. Dans un tel cas, on commence par traiter les cellules avec du formaldéhyde, qui provoque des pontages covalents entre les protéines et l'ADN. Comme nous le verrons un peu plus loin, ces pontages sont solides mais peuvent être renversés par la chaleur (67°C).
L'ADN génomique, maintenant ponté avec une grande quantité de protéines, est ensuite brisé pour donner des fragments de petite taille (environ 500 paires de bases). La technique la plus utilisée pour cette fragmentation est la sonication.
Pour récupérer les fragments d'ADN sur lesquels se trouve pontée la protéine qui nous intéresse, on utilise un anticorps spécifique contre cette dernière. L'anticorps sera idéalement biotinylé, ce qui permettra de le retenir contre une bille magnétique couplée à la streptavidine. Le complexe bille magnétique- streptavidine- anticorps- protéine pontée- ADN est retenu au fond d'un tube par un aimant pendant que tous les autres complexes ADN-protéines sont éliminés par lavage.
Dans la figure qui suit, on utilise un anticorps qui reconnait une protéine représentée en vert.
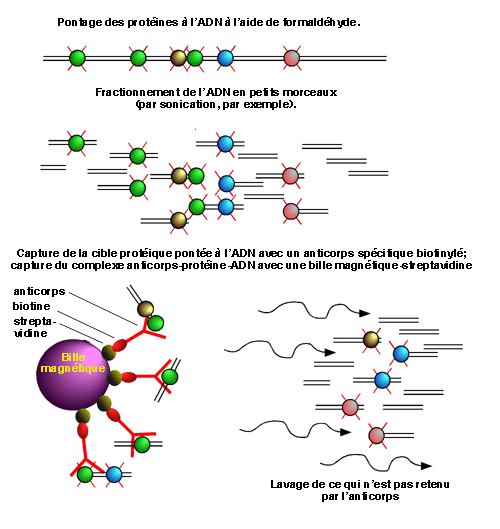
Après les lavages, la bille magnétique a permis de retenir tous les fragments d'ADN avec lesquels la protéine d'intérêt a une interaction assez importante pour permettre le pontage au formaldéhyde. Reste maintenant à identifier ces fragments..
On commence par chauffer le complexe à 67°C pour renverser le pontage au formaldéhyde. Cela permet la libération des fragments d'ADN, qui sont récupérés et purifiés. Bien sûr, ils sont en faible quantité et il faut ensuite les amplifier par PCR pour pouvoir établir leur présence.
La façon la plus simple de procéder est d'utiliser une paire d'amorces spécifiques à une région particulière (la région promotrice d'un gène qui nous intéresse, par exemple) et de procéder soit à un PCR normal, soit à un Q-PCR (aussi appelé "real-time PCR", qui permet d'effectuer des analyses plus quantitatives). Si la protéine étudiée était associée à la région qui nous intéresse, des fragments d'ADN contenant cette séquence auront été retenus lors des lavages et le PCR pourra en amplifier la séquence.
7.3.2 ChIP on chip ou ChIP2
Plutôt que de se limiter à une seule région lors de la réaction de PCR, on a aussi l'option (plus coûteuse!) d'analyser la présence de notre protéine partout dans le génome.
Pour ce faire, on commence par une ligation d'adapteurs aux extrémités des fragments d'ADN purifiés lors du ChIP. On utilise ces adapteurs pour pratiquer une réaction de PCR amplifiant tous les fragments présents. Cette amplification se fait en présence d'un nucléotide associé à une substance fluorescente, du genre Cy3 ou Cy5.
Pour identifier et démêler tous les fragments obtenus, on les hybride ensuite sur une micropuce d'ADN (aussi appelée "DNA chip", d'où le nom de "ChIP on chip"). Plus la micropuce contient de séquences, et plus celles-ci sont courtes, plus la résolution obtenue est élevée en termes de localisation des protéines dans le génome. (Bien entendu, le résultat sera plus intéressant si la micropuce contient non seulement des séquences codantes, comme c'est le cas pour les puces de cDNA, mais aussi des séquences intergéniques qui contiennent des régions de contrôle. L'idéal serait une puce portant des séquences couvrant le génome entier).
En utilisant une telle technique, si on dispose d'une micropuce adéquate, on peut (par exemple) suivre l'association globale d'un facteur de transcription à l'ADN pendant le cycle cellulaire, ou suite à un traitement quelconque, ou encore déterminer d'un seul coup d'oeil la position de plusieurs composantes de la chromatine dans différentes situations.
7.3.3 ChIPPET
Technique d'abord publiée dans Wei et al. (2006) Cell 124, 207-219.
Des données, des données, trop de données!!! C'est le drame des biologistes moléculaires modernes. Toute technique capable de réduire la quantité de bruit par rapport aux informations cherchées ne peut être que la bienvenue.

La technque du ChIPPET (Chromatin immunoprecipitation Paired End diTags) s'utilise pour les analyses à grande échelle, du genre "où sur les chromosomes trouve-t-on la protéine p53 quand on traite des cellules NIH3T3 avec de la caféine?" Elle utilise d'abord une immunoprécipitation comme on l'a vu ci-dessus, mais va ensuite remplacer l'étape d'hybridation sur une micropuce par une technique de séquençage. Oh, rassurez-vous, on ne va pas cloner chacun des fragments de chromatine immunoprécipités dans un vecteur pour ensuite les séquencer un par un! Ce serait bien entendu beaucoup trop long et finalement pas moins cher que d'utiliser une hybridation sur puce d'ADN.
Le ChIPPET essaie de résoudre deux problèmes associés à l'immunprécipitation de chromatine. Le premier est que la fragmentation de la chromatine par sonication donne des fragments de taille inégale (on vise ~500 pb, mais il s'agit là d'une valeur d'une moyenne). En outre, on immunoprécipitera toujours une certaine part de fragments par erreur; ce seront de faux positifs.
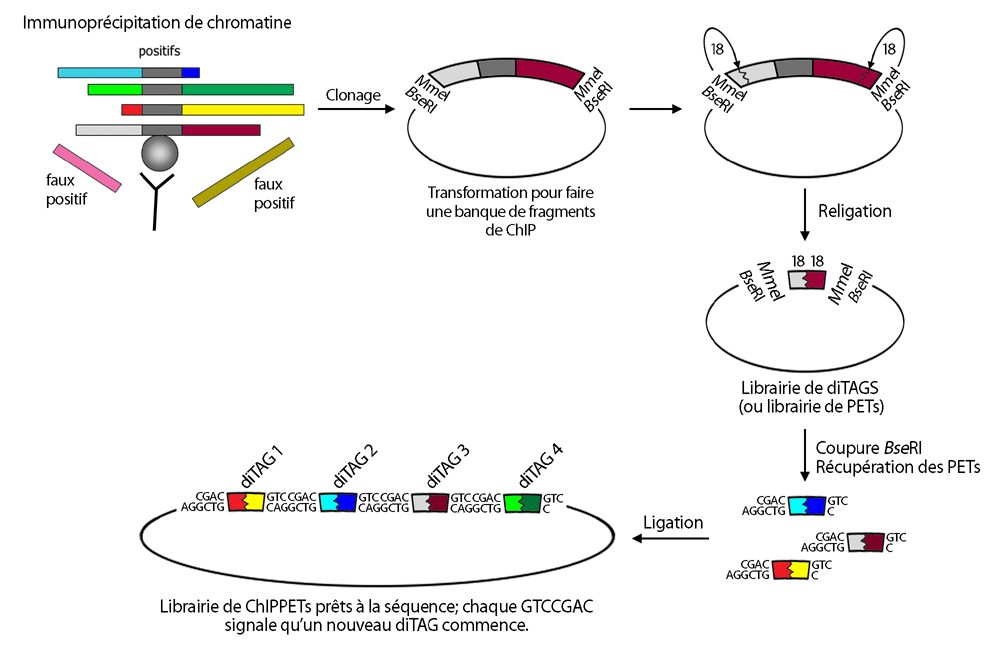
Après l'étape de ChIP, le ChIPPET demandera qu'on clone les fragments immunoprécipités dans des vecteurs entre deux sites Mme I. Cet enzyme coupe en dehors de son site de reconnaissance : TCCRAC,20,18. Le clonage de tous ces fragments donnera donc une banque de plasmides qu'on appellera une librairie de ChIP.
On va ensuite couper notre librairie de ChIP avec l'enzyme Mme I avant de refermer les plasmides avec de la ligase. Cette étape enlève la majeure partie des fragments immunoprécipités et n'en laisse que deux bouts par plasmide, soit les 18 pb en 5' de l'insert et les 18 pb en 3' de l'insert. Ces deux fragments de 18 pb recollés ensembles forment une étiquette (le diTag) qui est caractéristique d'une position dans le génome. La librairie de vecteurs ainsi coupés et religués est maintenant affublée du très flatulent nom de librairie de PETs.
La librairie de PETs est ensuite coupée avec un autre enzyme présent dans le site de clonage multiple, l'enzyme BseR I, qui lui aussi coupe en dehors de son site de reconnaissance (GAGGAG, 10,8). En raison de sa position par rapport au site Mme I du vecteur original, BseR I causera la formation de fragment d'environ 40 pb ayant cette allure générale :
CGAC - DiTAG de 36 paires de bases - GTC
AGGCTG - DiTAG de 36 paires de bases - C
On comprendra que chacun de ces inserts correspond à un fragment immunoprécipité, dont la position dans le génome peut être déterminée en cherchant par homologie de séquence un endroit où les deux TAGs de 18 pb seraient à environ 500pb l'un de l'autre.
Tous ces inserts sont ligués ensemble dans des vecteurs, formant une troisième librairie, la librairie de ChIPPET dont chaque composante sera séquencée séparément. Chaque réaction de séquence identifiera un grand nombre de fragment grâce à ses diTAGs.
Là où on gagnera en confiance dans les résultats, c'est en comparant les séquences obtenues. Des faux positifs seront rares alors que de vrais positifs seront plus abondant (ce qui est vrai dans la plupart des techniques). En outre, les faux positifs générés lors de l'amplification des différentes librairies se trahiront par leurs séquences identiques; ils auront tous les mêmes bouts. Ce problème peut aussi se manifester lors du ChIP utilisant le PCR; certaines séquences s'amplifient mieux que d'autres, donnant l'impression qu'elles ont été plus souvent immunoprécipitées. Ici, les vrais positifs donneront des diTags voisins mais pas identiques, puisque la sonication ne brise pas la chromatine de manière précise.
7.3.4 ChIPSeq
Cette technique basée sur la technologie de la compagnie Illumina vise à immunoprécipiter des fragments de chromatine et à en purifier l'ADN, puis à les séquencer directement sur une puce. Comment séquence-t-on de l'ADN collé sur une puce? C'est ce que nous allons voir.
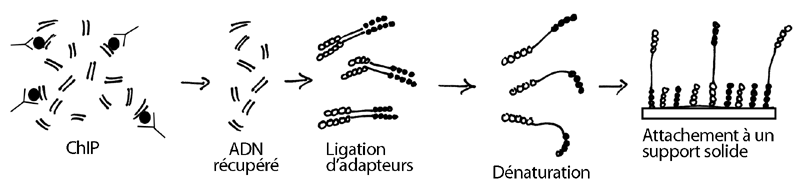
Supposons qu'on veuille séquencer le génome du biturong, un mammifère asiatique. À partir de cellules de l'animal, la technique demande qu'on en purifie l'ADN et qu'on le fragmente ensuite en petits morceaux en laissant des bouts francs. L'ADN passera par les étapes qui suivent :
1- Ligation d'adapteurs double-brin (A et B) aux deux bouts des fragments
2- Dénaturation de l'ADN et attachement des fragments simple-brin sur une surface solide ressemblant à une lame de microscope (grâce aux adapteurs, qui portent une modification à cet effet) . Les fragments s'attachent au hasard sur la surface de la plaque. Une très grande quantité d'adapteurs simple-brin (qui sont donc des amorces potentielles) se trouve aussi attachée sur le support solide, tout autour des plus grands fragments d'ADN.
3- Les longs fragments se replient et leur extrémité libre (qui porte un des brins de l'un des adapteurs) s'apparie aux courtes amorces complémentaires collées aur le support solide.
4- L'ajout de nucléotides ordinaires et d'ADN polymérase permet l'élongation des amorces qui auront réussit à s'annéliser sur les longs fragments, ce qui redonne un fragment double-brin.
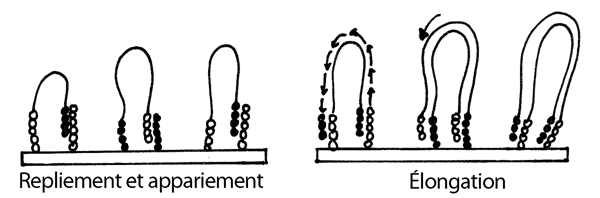
5- Dénaturation de ces fragments double-brin; chaque brin reste attaché par son amorce fixée au support solide.
6- Nouveau repliement des fragments simple-brin (maintenant deux fois plus nonbreux) et nouvel appariement aux amorces qui se trouvent en grande quantité dans le voisinage. Nouvelle élongation. On répète ce cycle plusieurs fois, jusqu'à ce que chaque fragment initialement collé au support donne naissance à un amas de fragments semblables.
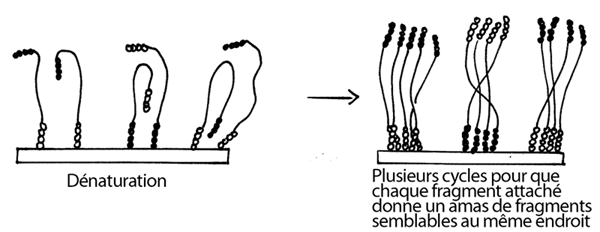
7- Ajout d'un nucléotide terminateur fluorescent, d'amorces et de polymérase. Un seul nucléotide est ajouté à chaque fragment, au bout de l'amorce de séquençage. L'appareil enregistre la position de chacun des points sur le support solide (ci-dessous je n'en ai dessiné que deux, mais il peut y en avoit plusieurs millions). Il identifie aussi le nucléotide qui a été ajouté à la position 1 pour chque point, en raison de sa couleur spécifique.
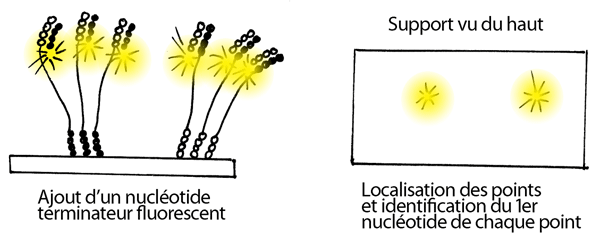
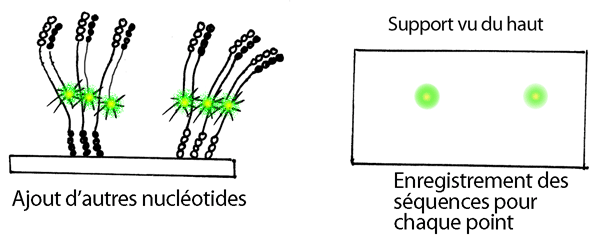
8- Les réactifs sont lavés, le groupement qui bloque le bout 3' du premier nucléotide est coupé chimiquement, le fluorophore du premier nucléotide est coupé chimiquement, et un deuxième nucléotide est rajouté. Comme ci-dessus, l'appareil enregistre quel nucléotide s'est ajouté à quel point. On répète ainsi les cycles base par base, en enregistrant la séquence de chacun des points.
7.3.5 ChIC & ChEC
Ces deux méthodes procèdent de la même idée, et augmentent la résolution des expriences de ChIP .
Le ChIC (Chromatin ImmunoCleavage) utilise une fusion entre la protéine A et la nucléase micrococcale (on l'appelle pA-MN). On procède à une fixation des protéines sur l'ADN comme dans un ChIP normal. On purifie ensuite les noyaux grossièrement, juste pour les rendre accessibles aux anticorps. Un premier anticorps est ajouté, visant la protéine dont nous voulons déterminer la position. Un deuxième anticorps (anti-lapin, anti-souris) est ensuite utilisé pour augmenter le recrutement de pA-MN au site occupé par le premier anticorps. La parite pA de pA-MN reconnaît et se fixe à la partie Fc des anticorps, ce qui amène sa partie MN dans le voisinage de l'ADN. L'ajout de Ca2+, un cofacteur essentiel de la nucléase micrococcale, permet alors à cette dernière de couper l'ADN très près du site de laison de la protéine étudiée. La résolution obtenue est de l'ordre de 100-200 pb, alors qu'un ChIP normal est plutôt du genre 500 pb.
Dans le ChEC (Chromatin Endogenous Cleavage), pendant ce temps, on produit directement une protéine de fusion entre la protéine d'intérêt et la nucléase micrococcale. En présence de calcium, cette protéine de fusion va couper l'ADN juste à côté de son site de liaison; la résolution en est augmentée encore plus.
Ces deux techniques ont été publiées par le laboratoire du Dr Laemmli (le même que celui du tampon éponyme!) dans Schmid et al. (2004) Cell 16, 147-157.
| Retour à la page d'accueil | Section précédente | Section suivante |