| Retour à la page d'accueil | Section précédente | Section suivante |
5.6.9 Chromatographie d'affinité
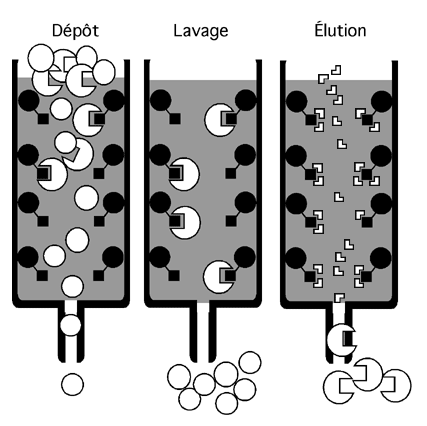
Dans une telle technique, on a recours à l'affinité de certaines molécules pour d'autres. On pourra ainsi fixer des anticorps sur une matrice solide dans une colonne (Protéine A-sépharose, par exemple) et faire passer notre échantillon sur la colonne pour y attraper les protéines reconnues par l'anticorps.
On a souvent recours à des colonnes dont la matrice porte des fragments spécifiques d'ADN pour aller pêcher les protéines liant l'ADN. Le bromure de cyanogène (CNBr) est l'un des réactifs utilisés pour coupler ainsi une cible à la matrice d'une colonne.
Plusieurs protéines de fusion ont des étiquettes permettant de reconnaître et de fixer des résines spécialement adaptées. Une telle étiquette est la glutathione-S-transférase, qui permet à la protéine à laquelle elle est fusionnée de se fixer sur une résine de glutathione-sépharose. La chitin-binding-protein (CBP) fait la même chose pour une résine couplée à de la chitine, un polymère de N-acétylglucosamine.
Les lectines comme l'agglutinine de germe de blé (WGA, pour wheat germ agglutinin) ont la très utile capacité de permettre l'adsorption de protéines glycosylées. Le WGA lie le N-acétylglucosamine; la concanavalin A (Con A) lie le mannose et le glucose
Puisque la chromatographie d'affinité repose sur l'interaction entre deux ou plusieurs molécules, on devine que seule l'imagination limite ses possibilités de combinaison. Il suffit de disposer de deux substances qui ont de l'affinité l'une pour l'autre et d'en fixer une sur un support solide.
On retrouve dans le commerce des résines d'affinité déjà faites. En voici quelques unes vendues par la compagnie Amersham.
|
Résine |
Affinité pour... |
|
Calmodulin sepharose 4B |
ATPases |
|
5' AMP sepharose 4B |
kinases dépendantes de l'ATP |
|
DNA-agarose |
ADN polymérases, protéines liant l'ADN |
|
Colonnes d'héparine |
Lipases, protéines liant l'ADN |
|
Poly(U) sepharose |
Transcriptase réverse, ARNm |
|
Arginine sepharose 4B |
Sérine protéases |
|
Lysine sepharose 4B |
ARNr |
|
Cibacron blue F3G-A |
enzymes nécéssitant des nucléotides |
|
ConA Sepharose 4B |
glycoprotéines contenant du a-D-mannose, du a-D-glucose et des sucres similaires |
|
Wheat germ Lectin Sepharose |
glycoprotéines contenant du N-acetyl-b-D-glucosmine |
On en retrouve d'autres sur le site de la compagnie.
Des résines "pré-activées" et prêtes à recevoir un ligand pour former une nouvelle résine d'affinité existent aussi.
|
Résine |
Groupe à coupler |
|
CNBr-activated Sepharose 4B |
-NH2 |
|
EAH Sepharose 4B |
-COOH |
|
Activated thiol sepharose 4B |
-SH |
|
rProtein A sepharose |
Partie Fab des anticorps |
5.6.10 IMAC (immobilized metal affinity chromatography)
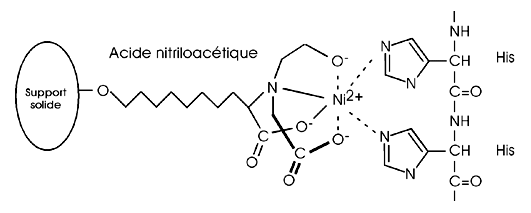
Ce type de chromatographie d'affinité fait appel à l'immobilisation, sur une résine, d'un atome métallique comme le nickel ou le cobalt. Cet atome immobilisé peut établir des liens de coordination avec certains polypeptides, comme par exemple une chaîne d'histidines. L'étiquette 6-His est justement ajoutée aux protéines pour que ces dernières soient plus aisément purifiées par chromatographie d'affinité pour le métal.
Dans la figure ci-dessous, une résine IMAC populaire (nickel- nitriloacetic acid ou nickel-NTA) utilise un ion de nickel comme site actif. L'ion Ni2+ possède six liens de coordination. Quatre sont sollicités par le NTA pour l'immobiliser sur la résine; il en reste donc deux pour interagir avec les atomes d'azote du cycle de la chaîne latérale de deux résidus histidines.
À pH <6, les résidus histidines commenceront à être réduits et ne pourront plus lier le nickel. On peut donc utiliser une variation de pH pour faire décoller les protéines. Cependant, la technique d'élution la plus usuelle (plus douce) se fera à l'aide d'un composé qui compétitionnera avec l'histidine pour le nickel: l'imidazole.
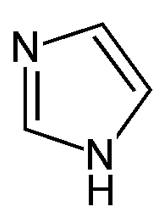 |
Imidazole, un composé compétitionnant avec l'histidine pour permettre l'élution de protéines d'une colonne d'affinité pour le métal immobilisé. |
Le nickel peut être chélaté par l'EDTA et l'EGTA, aussi ces composés doivent être utilisés avec parcimonie (et selon les instructions du fournisseur en résine). Les agents réducteurs peuvent aussi avoir un effet néfaste et doivent aussi être utilisées selon les instructions du manufacturier (qui a généralement amplement testé sa résine)!
Comme une étiquette multi-histidines est très petite, facilement clonable à l'extrémité N' ou C' d'une protéine, qu'elle permet la purification par affinité et peut être reconnue par des anticorps spécifiques disponibles commercialement, elle est un excellent choix de modification mineure facilitant la purification d'une protéine recombinante. Son aléa majeur est qu'il existe des protéines contenant naturellement des séquences répétées d'histidines; il faut donc se débarasser de celles-ci par une autre étape. (une recherche BLAST sur le protéome de S. cereviasiae révèle 15 protéines avec au moins six histidines d'affilée). Pour de telles protéines, une colonne IMAC peut servir de colonne d'affinité même sans l'ajout d'une étiquette.
5.6.11 Système de chromatographie
La figure ci-dessous décrit un système de base de chromatographie (il est inspiré du système FPLC d'Amersham Biosciences).
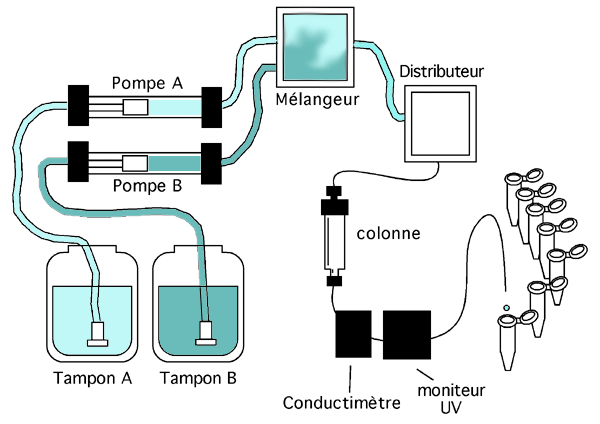
Le coeur de ce système est évidemment la colonne. Elle est reliée au reste du système de façon à ce que le tampon la traverse de haut en bas. Ce tampon provient de réservoirs situés tout au bout en amont du système. On a ici représenté deux tampons différents (A et B) reliés chacun à une pompe (A et B), même si bien entendu il est possible de faire fonctionner ce système avec un seul tampon et une seule pompe. L'avantage d'en avoir deux est qu'on peut programmer le système pour pomper progressivement plus d'un tampon que de l'autre, générant ainsi un gradient. (D'ordinaire, les deux tampons ont la même composition sauf pour la concentration en sel; faire varier la proportion de l'un par rapport à l'autre pendant la chromatographie génère un gradient de force ionique dans la colonne).
Chaque pompe est reliée au mélangeur, une chambre où s'effectue le mélange des deux tampons. Le tampon de chromatographie passe ensuite dans le distributeur, qui l'enverra soit vers la colonne, soit vers une autre destination. C'est aussi dans le distributeur qu'on déposera l'échantillon à chromatographier; le distributeur le dirige vers la colonne.
En aval de la colonne, on installe d'habitude un conductimètre, qui enregistre la conductivité du tampon (et donc sa concentration en sel) et une cellule spectrophotométrique qui enregistre l'absorbance aux UV pour détecter le passage des protéines. L'éluat de la colonne est ensuite recueilli en petites fractions par un collecteur.
La figure ci-dessous nous montre ce à quoi ressemble l'enregistrement de la densité optique de l'éluat en fonction du temps pendant une chromatographie d'échange d'ions avec un gradient de sel.
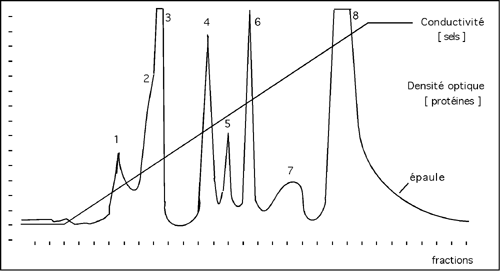
Le conductimètre a enregistré le passage d'une concentrations en ions de plus en plus élevée avec le temps. Les protéines, elles, décrochent de la colonne en fonction de l'effet qu'a ce gradient de sel sur leur interaction avec les groupements chargés de la colonne. Les protéines du pic 1 décrochent avant les protéines du pic 8; c'est donc que leur interaction avec la colonne était plus faible.
Remarquez que les pics ont des formes différentes. La plupart d'entre eux reflètent sûrement la présence de plusieurs protéines différentes, surtout si cette chromatographie a été pratiquée en début de purification. On remarquera aussi que certains pics sont fusionnés: ainsi, le pic 2 court presque à la même place que le pic 3 et lui donne une épaule. On trouve une autre épaule, beaucoup plus importante, dans le pic 8; cependant, dans ce cas, l'épaule peut simplement être due à la très grande taille du pic.
5.6.12 HPLC
High performance liquid chromatography: il s'agit d'une technique permettant de séparer les protéines (entre autres) avec une très grande résolution.
En gros, un système HPLC ressemble au système vu ci-haut: les différences majeures seront que la colonne est généralement en acier et que le tampon de chromatographie (le solvant) est sous haute pression pendant la séparation.
Plusieurs types de séparation sont possibles en HPLC. La chromatographie d'échange d'ions et la filtration sur gel s'y pratiquent selon les mêmes principes qu'avec une chromatographie "low pressure" (même si à strictement parler, ce n'est pas un "gel" qu'on trouve ici dans la colonne pour effectuer la filtration).
On distingue souvent en HPLC la chromatographie en phase normale de la chromatographie en phase inverse. En phase normale, la matrice de la colonne est très polaire (de style silice, par exemple) et la phase mobile (le solvant) est non-polaire (n-hexane, par exemple). En phase inverse, vous l'avez deviné, c'est le contraire: la matrice de la colonne est non-polaire ou hydrophobique et le solvant est polaire (eau, méthanol, acétonitrile). C'est le pendant à haute pression de la chromatographie à interaction hydrophobe.
Le HPLC se distingue par le très petit diamètre de ses colonnes (moins de 5 mm); un flot à haute pression de solvant; la capacité de séparer et détecter de très petites quantités; et finalement une très haute résolution.
Sur le sujet du HPLC, je vous recommande le livre en ligne des Drs Yuri Kazakevich et Harold McNair.
5.6.13 Techniques de précipitation
On a souvent recours à une précipitation d'un aliquot de notre préparation pour des fins analytiques (pour mettre les protéines sur gel, par exemple). Il existe plusieurs techniques rapides et fiables pour précipiter les protéines. Je recommanderais de se tenir loin de toutes celles qui utilisent le triethanolamine, cependant, à cause de son odeur de latrines de métro.
Voici deux protocoles rapides et efficaces ne demandant que très peu de manipulations. Attention à vos doigts: ils sont couverts de kératine qui va promptement contaminer vos échantillons si vous n'y prenez garde! Le port de gants de laboratoire est de rigueur.
|
Précipitation des protéines à l'acide trichloroacétique
1- ajuster les protéines à 25% TCA (trichloroacetic acid) 2- Laisser précipiter 30 minutes sur glace 3- Centrifuger 10 minutes (microfuge, 14 000 rpm) 4- Laver avec 300 uL de 5mM HCl dans l'acétone (au vortex) 5- Centrifuger 5 minutes 6- Laver à l'acétone pure (vortex) 7- Centrifuger 5 minutes 8- Les protéines sont dans le petit culot. |
|
Précipitation des protéines au méthanol 1- À 100 uL de protéines, ajouter 400uL de méthanol (vortex). 2- Ajouter 100 uL de chloroforme (vortex) 3- Ajouter 300 uL H2O (vortex) 4- Centrifuger 2 minutes (microfuge, 14 000 rpm) 5- Les protéines sont à l'interface. Retirer la phase supérieure et jeter. 6- Ajouter 300 uL de méthanol au reste du mélange. 7- Centrifuger 2 minutes (microfuge, 14 000 rpm) 8- Les protéines sont dans le petit culot. |
5.6.14 Pontage
Les protéines s'associent souvent à d'autres molécules. Il est indispensable parfois de pouvoir préserver ces associations pendant une purification. Bien qu'il soit parfois possible d'utiliser certaines méthodes douces préservant les associations en questions, très souvent on doit s'assurer qu'elles tiennent bon en les aidant un peu à l'aide d'un traitement chimique.
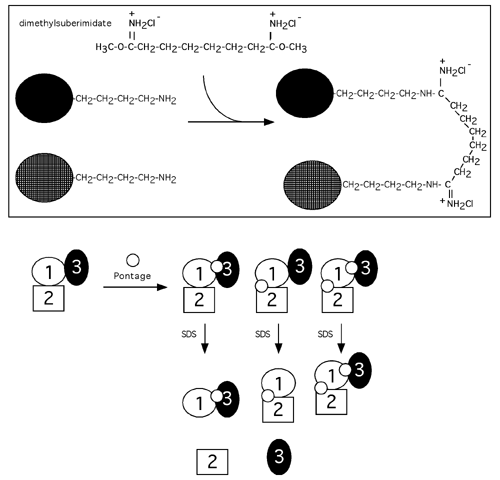
Un exemple de pontage entre les sous-unités d'une protéine est donné ci-dessous. Dans ce cas on a eu recours au DMS, ou diméthylsuberimidate. (Ne pas confondre avec un autre DMS, le diméthylsulfate ou encore avec le DMSO, le diméthylsulfoxide). Il est possible que toutes les sous-unités ne soient pas pontées en même temps, mais à l'analyse on peut déduire la nature de leurs interactions.
Ici, comme on découvre sur gel SDS des bandes correspondant à des complexes des sous-unités 1-2-3, des sous-unités 1-2 et 1-3 mais pas des sous-unités 2-3, on pourrait conclure que la sous-unité 1 contacte les deux autres dans l'holoenzyme, mais que les sous-unités 2 et 3 n'ont pas de contact direct.
| Retour à la page d'accueil | Section précédente | Section suivante |