BCM-514
Biochimie des protéines
5.0 La purification des protéines
5.5 Extraction
5.5.1 Généralités
Quelque soit le type de cellule que nous ayons choisi pour produire la protéine qui nous intéresse, nous devrons persuader cette dernière d’en sortir. À moins que notre candidate ne soit une protéine sécrétée et donc disponible dans le milieu, nous devrons sélectionner une méthode pour briser les cellules et en extraire le contenu.
Plusieurs choix s’offrent à nous, choix qui seront faits en fonction du type de cellule utilisé, des conditions de purification de notre protéine, de l’équipement disponible, et même de nos goûts personnels. Soyons conscients de ce que certaines méthodes d’extraction seront clairement préférables à d’autres même si elles sont plus compliquées; il nous faut donc être souples quant à notre décision finale.
Les principales approches de lyse des cellules sont les suivantes. Un choc osmotique peut suffire à briser la membrane cellulaire de cellules fragiles. Ce choc peut être assisté par un léger traitement avec un détergent (ce qui permet aussi d’aider à maintenir les protéines en solution). Une certaine action mécanique peut aussi être ajoutée au processus: un homogénéisateur à piston permet de briser la plupart des cellules de mammifères.
Pour les cellules de plantes, de levures ou les bactéries, toute approche "douce" devra tenir compte de la présence d’une paroi cellulaire très résistante; les protoplastes de telles cellules seront préparés en traitant ces dernières avec des enzymes (telle la lyticase) qui détruiront la paroi sans briser la membrane plasmique.
Finalement, dans tous les cas, une méthode brutale peut aussi être utilisée. Le traitement aux ultrasons, une lyse mécanique utilisant des billes de verre ou une presse Aminco-French, comme les cycles de congélation/décongélation, sont tous des moyens envisageables.
5.5.2 Tampon
L’extraction s’effectuera dans une solution tampon, dans laquelle les cellules seront resuspendues après une centrifugation visant à les concentrer. À cette étape, notre protéine est fort probablement en concentration infime par rapport à toutes les autres protéines cellulaires; nous mettrons les chances de notre côté en limitant toute dilution supplémentaire. On cherchera autant que possible à maintenir une concentration élevée dès les premières étapes.
Le tampon de lyse répondra à certaines exigences expérimentales. Il devrait avoir un fort pouvoir tampon, car le contenu des cellules peut être à un pH inapproprié. On ne veut pas, par exemple, que les enzymes lysosomiques puissent se retrouver tout d’un coup libérés de leur enveloppe et dans un milieu à pH acide, conditions dans lesquelles ils auraient tout loisir de détruire les autres protéines cellulaires.
Rappelons-nous que le pouvoir tampon d’une substance est plus élevée dans une région de pH proche de son pKa; voici les pKa de certains tampons populaires.
|
Bis-TRIS |
6,46 |
|
PIPES |
6,76 |
|
MOPS |
7,20 |
|
TES |
7,40 |
|
HEPES |
7,48 |
|
Tris |
8,06 |
|
Glycine |
9,78 |
Le HEPES (pour la chromatographie d’échange de cations) et le Tris (pour l’échange d’anions) sont ceux que vous rencontrerez le plus souvent en purification de protéines. Méfiez-vous des solutions "tampon" qui ont un pH très éloigné du pKa; à plus de deux ordres de grandeur en plus ou en moins que le pKa, ce ne sont sûrement pas de très bons tampons.
Alors que l’intérieur des cellules est un environnement réducteur (à cause entre autres de toute cette glutathione), l’atmosphère dans laquelle nous vivons est au contraire oxydante (elle est riche en oxygène). Il y a gros à parier que notre protéine devra être protégée d’une oxydation excessive : c’est pourquoi un agent réducteur comme la glutathione, l’acide ascorbique, le beta-mercaptoethanol ou le dithiothreitol (DTT) pourra être ajouté au tampon. Ne vous en faites pas trop pour les ponts disulfures retrouvés à l'intérieur des protéines globulaires: ils devraient être bien à l'abri des conditions externes.
On retrouve souvent du glycérol dans la composition des tampons. Il stabilise les interactions protéine-protéine.
Faisons face à cette dure réalité : notre protéine a beaucoup d’ennemis. La cellule même d’où elle provient est remplie de protéases qui ne demandent pas mieux que d’en faire une bouchée. En brisant les lysosomes (particulèrement si nos cellules de départ ont une forte activité catabolique, comme les cellules du foie; ou si ce sont des cellules de plantes avec des vacuoles riches en protéases) nous avons libéré une horde de protéases féroces dans le même tampon de lyse que notre protéine. La solution? Tenir le pH élevé (entre 7 et 8), garder la soupe au frais (4°C) et ajouter des inhibiteurs de protéases à la solution.
Voici quelques-uns de ces inhibiteurs :
|
Inhibiteurs |
Cible
|
|
PMSF |
Toutes les sérine protéases (trypsine, thrombine, chymotrypsine, papaine, etc). |
|
AEBSF |
Toutes les sérine protéases (trypsine, thrombine, chymotrypsine, papaine, etc). Plus soluble et moins toxique que son prédécesseur, le PMSF. |
|
EDTA et EGTA |
métalloprotéases (métal comme cofacteur) |
|
Benzamidine |
sérine protéases |
|
Pepstatin A |
protéases aspartiques comme la cathepsine D, la rénine, la pepsine et les protéases d'HIV. |
|
Leupeptin |
sérine- (trypsine, plasmine, kallikréine porcine) et cystéine- protéases (papaine, cathepsine B). N'inhibe pas la thrombine ni la chymotrypsine. |
|
Aprotinin |
sérine protéases, mais pas la thrombine ou le facteur X. |
|
Chymostatin |
sérine protéases avec une spécificité de type chymotrypsine (chymotrypsine, chymases, cathepsine G et des cystéine protéases comme les cathepsines B, H et L. |
|
Antipain |
Inhibe la papaine, la trypsine et la plasmine jusqu'à un certain point. Plus spécifique pour la trypsine et la papaine que ne l'est la leupeptine. |
Notez que l’acétylcholine estérase est une sérine protéase; la bungarotoxine du cobra pourrait donc être utilisée comme agent protecteur de votre protéine. Si vous cherchez un animal familier dans le labo, optez donc pour un cobra. Il s’occupera aussi des souris qui s’échappent de leur cage.
Plusieurs compagnies offrent un catalogue très complet d’inhibiteurs de protéases, de même que des cocktails déjà mélangés.
5.5.4 Techniques d’extraction
Si du tissu entier est choisi comme matériel, surtout s’il est riche en tissu conjonctif, il faudra d’abord libérer un maximum de cellules. Un blender de cuisine sera un outil fort utile pour ce faire, malgré son origine infâme pour les compagnies d’outillage de laboratoire. On utilisera aussi un enzyme comme la collagénase pour briser les fibres de collagène empêtrant les cellules.
5.5.4.1 Choc osmotique
Le choc osmotique permet de briser certaines cellules fragiles avec un minimum de dommages. Les cellules sont incubées dans une solution hypo-osmotique. Cherchant à rétablir l’équilibre osmotique, l’eau pénètre dans la cellule et finit par causer une rupture de la membrane plasmique. On peut donner un coup de pouce à la technique avec un peu de détergent (NP-40, LDAO, triton X-100) et avec un soutien mécanique (quelques coups de piston dans un homogénéisateur de type Dounce). Une telle approche permet de récupérer des noyaux presque intacts une fois séparés des débris de la membrane plasmique.
Il est possible de lyser les membranes plasmiques sans briser les noyaux. La lyse cellulaire doit être suivie par de fréquentes visites aux microscope. La lyse des noyaux, si elle arrive accidentellement, se traduira par une soudaine augmentation de la viscosité de la solution: c'est la chromatine qui est libérée qui en la responsable.
Soyons francs : ce genre de technique relève de l’art autant que de la science et il n’y a rien de tel que la pratique répétée pour que l’expérimentateur en découvre toutes les nuances.
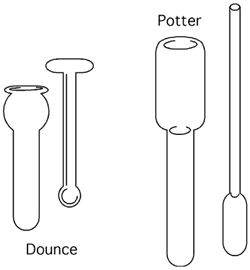
5.5.4.2 Homogénéisateur de type Dounce
|
Cet appareil ressemble à une éprouvette (aux parois épaisses) dans laquelle s’enfonce un piston serré. Le passage des cellules dans l’espace extrêmement exigu entre le piston et la paroi interne du tube cause leur bris, un peu comme ce qui nous arriverait si nous étions accroché à une porte de métro quand il s’engouffre dans un tunnel. Sproutch.
5.5.4.3 Homogénéisateur de type Potter-Elvehjem
Le "potter" ressemble à un Dounce motorisé; son piston a souvent une tête de teflon et tourne dans le cylindre contenant les cellules resuspendues.
|
|
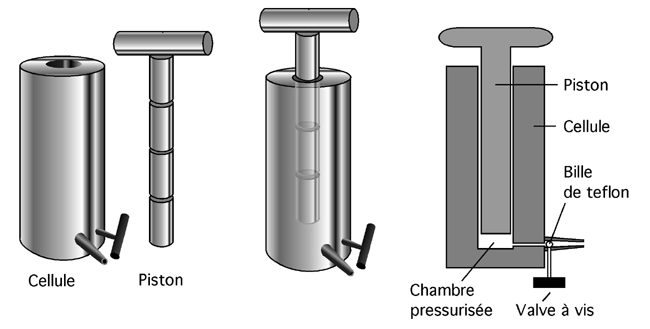
5.5.4.4 La presse Aminco- French

|
Ne vous laissez pas abuser par son sobriquet anglais de French press : cet appareil n’est pas français, il a été mis au point par un certain Dr. French (un nom d’inventeur plus facile à porter, avouons-le, que Poubelle ou Crapper). L’appareil vise lui aussi à forcer les cellules à passer dans un espace plus petit qu’elles, ce qui déchire leur membrane (et réduit la plupart de leurs structures en marmelade). Il consiste en un cyclindre creux en métal, cylindre dans lequel s’enfonce un piston de métal nanti de plusieurs o-rings d’un caoutchouc très solide. À la base du cyclindre, une petite valve est installée; celle-ci peut être obstruée par une petite bille en teflon.
|
La suspension de cellules est versée dans le cyclindre. Le piston est installé, obstruant le trou, et une puissante vis sans fin commence à l’enfoncer dans le cylindre (avec un bruit assez inquiétant il faut bien l’admettre). Comme il n’y a que peu d’air dans le cyclindre et qu’un liquide est à toute fin pratique incompressible, l’enfoncement du piston fait très rapidement grimper la pression sur les parois du cylindre. Quand on ouvre légèrement la valve, la suspension de cellules est éjectée comme de l’eau qu’on fait passer entre ses dents et les cellules sont déchiquetées. Plus la pression est haute dans le cylindre, plus totale est la lyse.
5.5.4.5 La bombe à disruption
Elle consiste en une chambre pressurisée dans laquelle les cellules sont traitées avec de l'azote à haute pression. La pression force l'azote à se solubiliser dans les liquides. On libère alors la pression tout d'un coup; l'azote en solution reprend son état gazeux, forme des bulles à l'intérieur des cellules et les fait éclater.
C'est un problème similaire qui attend les plongeurs sous-marins qui remontent sans avoir respecté leurs paliers de décompression.
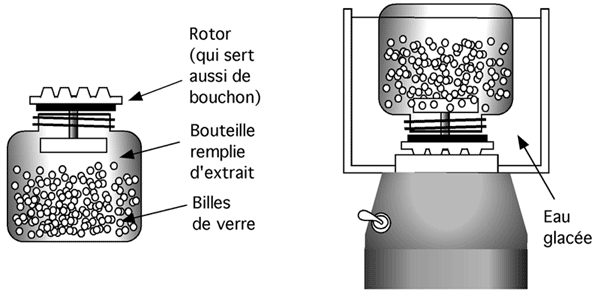
5.5.4.6 Les billes de verre
L’appareil utilisé pour cette technique ressemble à un blender (et un coup parti dans les anglicismes, pourquoi ne pas aller jusqu'au bout... on appelle ça un bead beater), à ceci près qu’il est beaucoup plus petit et ne contient pas de lames. Son contenant amovible est rempli de petites billes de verre sur lesquelles on verse la suspension de cellules. La suspension se répartit entre les billes. Il est important de remplir complètement le contenant et de ne pas y laisser d’air pour éviter de faire de la mousse et d’oxyder les protéines. Le tout est alors scellé et on lance la machine, qui fait furieusement tourbilloner les billes de verre dont l’action abrasive a tôt fait de réduire les cellules en charpie.
Séparer les billes du lysat est très facile parce que les billes coulent au fond dès que l'agitation cesse.
On doit prendre soin de garder le tout au frais; en fait le contenant est souvent nanti d’une chambre où on installe de la glace. Le mouvement des billes génère en effet beaucoup de chaleur.
5.5.4.7 La sonication
Elle consiste en la destruction des cellules par les ultrasons, exactement comme le faisait l’invention du professeur dans "L’affaire Tournesol" de Hergé. Ici, une tige de métal (le "sonicateur") à l’extrémité très fine est introduite dans la suspension de cellules et induite à vibrer violemment, émettant un bruit fort déplaisant même pour l’utilisateur portant ses cache-oreilles obligatoires.
Les vibrations sont des ondes de pression ultrasoniques qui causent la croissance de bulles par un phénomène appelé "cavitation". Ces bulles sont soumises à un grand stress par les ondes ultrasoniques et peuvent subitement grandir, se contracter ou même imploser. Une telle implosion est accompagnée d'une très haute augmentation locale de la pression et de la température. Quel genre d'augmentation? Et bien une microbulle qui implose ainsi peut voir sa température grimper à 5000°C et sa pression atteindre plusieurs centaines d'atmosphère.
Comment les microbulles se forment-elles? Une analogie possible est celle d'un attache-trombone qu'on tortille. Il est fait de métal, mais en le pliant et le dépliant rapidement vous pouvez faire augmenter sa chaleur, "fatiguer" les liens moléculaires qui assurent sa cohésion locale et le briser. C'est la même chose qui arrive aux molécules d'eau quand un sonicateur les fait vibrer. Les vibrations ultrasoniques réussissent à briser les ponts hydrogènes qui assurent l'état liquide de l'eau, ce qui les convertit en gaz. Il s'agit en quelques sorte d'une ébullition à froid, effectuée en agitant les molécules d'eau. Mais comment maintenir de l'eau à l'état gazeux dans un liquide froid? Assurément cela ne peut pas durer bien longtemps et le gaz se condense. Or voilà: le gas qui se condense perd du volume et laisse derrière lui un espace vide (une cavité, d'où le nom de "cavitation"). L'eau liquide se précipite dans l'espace vide pour le remplir, et les molécules d'eau liquides entrent en collision au coeur de la cavité. L'onde de choc de cette collision s'étend alors dans le voisinage comme les cercles concentriques générés par un caillou lancé dans une mare. Et l'addition de toutes ces ondes de choc peut pulvériser les cellules, comme le ferait un très puissant ouragan qui tomberait sur notre maison à l'improviste.
Le hic avec le sonicateur est qu'il génère beaucoup de chaleur dans le matériel biologique. L'utilisateur a tout intérêt à garder le tube dans lequel a lieu la sonication dans un bac à glace.
5.5.4.8 Enzymes lytiques
Avec les levures, les plantes, les bactéries, il faut tenir compte de la paroi cellulaire qui protège la membrane plasmique. Si on n’opte pas pour une méthode mécanique et violente d’extraction, il faudra d’abord détruire cette paroi avant de s’en prendre au reste de la cellule.
Différents enzymes comme le lysozyme (du blanc d'oeuf de poule, par exemple) ou la lyticase de S.aureus sont disponibles pour s’occuper de cette tâche; il suffit de choisir le plus approprié.
5.5.4.9 Élimination précoce de ce qui n'est pas utile
La cellule contient une très grande quantité de protéines, parmi lesquelles celle qui nous intéresse est fort probablement très minoritaire. Si possible, il sera donc tout à notre avantage de nous débarasser le plus vite possible de la majorité des protéines qui ne nous intéressent pas.
De nombreuses protéines sont concentrées dans une région limitée de la cellule : facteurs de transcription et histones dans le noyau, par exemple; protéines mitochondriales dans les mitochondries, etc. Plusieurs protocoles existent pour isoler ces organelles les unes des autres par centrifugation. Une extraction douce des organelles suivie de leur isolation nous donnera une très grande longueur d’avance pour la suite de la purification.
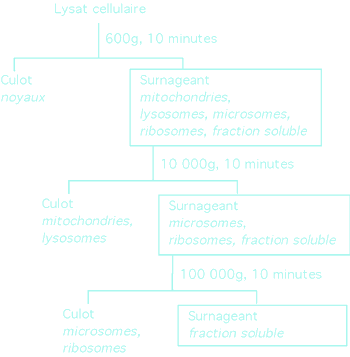
|
Un exemple de séparation des composantes cellulaires par centrifugation différentielle. Certaines organelles de la cellule sont plus denses que d'autres et plus à même de sédimenter à basse vitesse de centrifugation.
Si notre protéine est contenue dans une organelle particulière, nous pouvons gagner beaucoup de temps et d'effort en séparant d,abord celle-ci du reste des composantes cellulaires; c'est particulièrement le cas des noyaux.
|
Les protéines nucléaires sont presque toujours préparées à partir de noyaux et pas de cellules entières. Une bonne façon de séparer les noyaux du reste est de déposer le lysat cellulaire (avec les noyaux intacts) sur un coussin de sucrose. La densité du sucrose ne laisse passer que les particules denses comme les noyaux, les débarassant de la majeure partie du reste du matériel cellulaire.
La compagnie Caprion, de Montréal, s’est fait une spécialité de purifier les organelles. Une visite sur son site web www.caprion.com est fort instructive.
5.5.4.10 La chromatine
Selon la technique utilisée, nous nous retrouverons avec un extrait protéique brut contenant de l’ADN plus ou moins intact et associé à un grand nombre d’histones. Plus cette chromatine est intacte, et plus notre soupe est visqueuse. Une extraction à la presse Aminco-French ou au sonicateur va pulvériser la chromatine et n'en laisser que des débris; une extraction de protéines ne lysant pas les noyaux laissera la majeure partie de la chromatine derrière nous.
On peut séparer la plupart des protéines de la chromatine en changeant la force ionique de la solution (ce qui se passe lors d'une précipitation au sulfate d’ammonium, par exemple). L'histone H1 va décrocher à partir de 100 mM d'ammonium, ce qui aide la solubilisation de l'ADN; les histones de l'octamère décrochent entre 400mM et 700mM d'ammonium). On peut même utiliser une simple ultracentrifugation après la lyse nucléaire pour se débarasser de la chromatine (la chromatine intacte est un énorme réseau très visqueux qui s'écrasera dans le culot, alors que la plus grande partie des protéines solubles resteront dans le surnageant).
Mieux vaut se débarasser de l’ADN dès que possible.
On peut opter pour un traitement aux nucléases (si on ne craint pas de traîner des nucléases derrière nous) ou encore choisir comme première étape de purification chromatographique un passage sur une résine de DEAE, chargée positivement, qui retiendra fortement l’ADN chargé négativement. Ce serait en tout cas mon choix.
5.5.4.11 Protéines membranaires
Ces protéines ne se promènent pas librement en milieu aqueux puisqu’elles comptent au moins un domaine transmembranaire hydrophobe. Elles sont donc assez insolubles et difficiles à préparer. Leur purification requiert l’utilisation de détergents.
5.5.4.12 Corps d’inclusion
La même chose est vraie de protéines exprimés chez E.coli, surexprimées ou non, et qui se retrouvent dans des agrégats insolubles appelés corps d’inclusion. Notons au passage que la solubilité d’une protéine surexprimée chez E. coli peut varier selon les étiquettes qu’on y ajoute par ingénierie génétique; ainsi, il est possible qu’une même protéine soit insoluble avec une étiquette 6-histidines et soluble avec une étiquette GST.
Les corps d’inclusion peuvent être récupérés par centrifugatrion. Si nous ne craignons pas de travailler avec du matériel qui a précipité et a été resuspendu, nous pouvons alors entreprendre de resolubiliser les protéines qu’ils contiennent. Des agents vigoureux comme la guanidine-HCl, le SDS ou l’urée 8M sont utilisés pour resuspendre les agrégats; ils doivent ensuite être éliminés pour permettre la renaturation.
Le problème au point de vue expérimental est de jongler entre des conditions assez dénaturantes pour prévenir l'aggrégation et la précipitation, mais aussi assez douces pour ne pas systématiquement dénaturer toutes nos protéines. Chaque protéine demandera un protocole optimisé.
Voici le protocole du Protein Purification and Expression Facility de l'EMBL, à Heidelberg en Allemagne:
Tampon de lavage
50 mM Tris-HCl pH 7.5
50 à 200 mM NaCl
Tampon d'extraction
50 mM Tris-HCl pH 7.5
8 M urea
1 mM DTT
1 mM PMSF* * Le PMSF est instable en solution aqueuse et est ajouté au
tampon au moment décrit dans le protocole.
Procédure
- 1. Centrifuger le lysat cellulaire à 4°C pour 20 min. à 30 000 g (16 000 rpm dans un rotor SS34 de Sorvall ou JA-20 de Beckman).
- 2. Resuspendre le culot dans 10 mL de tampon de lavage contenant 1% triton X-100 et 1M urée par gramme de culot de cellules. Incuber à T/P pour 5 minutes.
- 3. Centrifuger le lysat cellulaire comme à l'étape (1).
- 4. Répéter 3 fois.
- 5. Resuspendre le culot dans 10 mL de tampon de lavage et centrifuger la suspension à 4°C pour 30 min. à 45 000 rpm dans un rotor 45Ti de Beckman.
- 6. Resuspendre les corps d'inclusion dans le tampon d'extraction. Utiliser assez de tampon pour obtenir une concentration finale en protéine de 1 mg/mL.
Ajouter 10 uL PMSF (100 mM) par mL de solution. Incuber une heure à T/P.
- 7. Diluer la solution dix fois avec le tampon d'extraction et dialyser toute la nuit contre 100 volumess de solution de lavage.
- 8. Centrifuger le dialysat à 4°C pour 30 min. à 45 000 rpm dans un 45Ti.
En voici un autre, de Laurent Vuillard et Alasdair Freeman de l'université de St Andrews (Royaume Uni).
Solubilisation des protéines des corps d'inclusions
- Resuspendre le culot d'un litre de culture batérienne dans 20 mL de
- 50mM HEPES-NaOH pH7.5
- 0.5M NaCl
- 1mM PMSF
- 5mM DTT
- avec 0,35mg/mL de lysozyme
Incuber 30 min. à 20°C.
- - Ajouter du triton X-100 à une concentration de 1% (vol./vol.)
- - Soniquer par coups de 30 secondes jusqu,à ce que la solution s'éclaircisse.
- - Traiter l'extrait à la DNAse I (20 mg/L) pour 60 min. à 37°C
- - Sédimenter les corps d'inclusion par centrifugation à 30 000g, 30 min., 4°C.
- - Laver le culot (les corps d'inclusion) deux fois au TBS ou au PBS contenant 1% Triton X-100, et centrifuger à 30 000g, 30 min., 4°C
- - Solubiliser le culot dans 2 mL de
- 50mM HEPES-NaOH pH7.5,
- 6M guanidine HCl,
- 25mM DTT
et laisser 1h at 4C.
- Le matériel insoluble est retiré par centrifugation à 100 000g pour 10 minutes. Cette étape est particulièrement importante pour enlever les aggrégats qui pourraient servir de noyaux de précipitation pendant le relpiement des protéines resolubilisées.
- Évaluer la concentration en protéines et ajuster à 1mg/mL en utilisant
- 50mM HEPES-NaOH pH7.5,
- 6M guanidine HCl,
- 25mM DTT
Repliement
Les protéines resolubilisées sont diluées aussi vite que possible 1 : 10 dans le tampon de repliement à 4°C:
- 50mM HEPES pH7.5,
- 0.2M NaCl,
- 1mM DTT,
- 1M NDSB256 (Phorbol 12,13 diacetate).
La concentration finale en protéines ne devrait pas excéder 0,05 à 0,1 mg/ml. Le mélange doit être fait rapidement. Pour de très petits volumes, on peut directement déposer la suspension protéique dans la solution de repliement en vortexant. Pour des volumes plus grands, plutôt qu'un vortex, utiliser un agitateur magnétique vigoureux. Agiter 2 minutes après l'addition. Incuber 1 heure à 4°C. Le reste du NDSB et de la guanidine peuvent être retirés par dialyse.
Notes
- Les paramètres-clés de ce protocole sont la concentration protéique, celle du guanidium résiduel et la température. Optimiser au besoin.
- Les NDSB (Non-detergent sulfobetaines) sont des agents aidant le repliement des protéines. Ce sont des zwitterions qui contiennent un groupement sulfobetaine hydrophilique et une courte chaîne hydrophobique; ils ne peuvent cependant pas former de micelles. Ils aident à réduire l'aggrégation et facilitent le repliement des protéines dénaturées (on présume que c'est en interagissant avec les groupements hydrophobes des protéines et en masquant ceux-ci à des groupes similaires sur d'autres protéines).
Le NDSB 256 est d'habitude un peu plus efficace que le NDSB 201 mais ce dernier est beaucoup moins cher.
benoit.leblanc@usherbrooke.ca